the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 27 Nov 2023 | OAE Guide 2023 | Chapter 3
| 27 Nov 2023 | OAE Guide 2023 | Chapter 3
Assessing the technical aspects of ocean-alkalinity-enhancement approaches
Matthew D. Eisaman
Phil Renforth
Laura Bastianini
James Campbell
Andrew W. Dale
Spyros Foteinis
Patricia Grasse
Olivia Hawrot
Carolin R. Löscher
Greg H. Rau
Jakob Rønning
Ocean alkalinity enhancement (OAE) is an emerging strategy that aims to mitigate climate change by increasing the alkalinity of seawater. This approach involves increasing the alkalinity of the ocean to enhance its capacity to absorb and store carbon dioxide (CO2) from the atmosphere. This chapter presents an overview of the technical aspects associated with the full range of OAE methods being pursued and discusses implications for undertaking research on these approaches. Various methods have been developed to implement OAE, including the direct injection of alkaline liquid into the surface ocean; dispersal of alkaline particles from ships, platforms, or pipes; the addition of minerals to coastal environments; and the electrochemical removal of acid from seawater. Each method has its advantages and challenges, such as scalability, cost effectiveness, and potential environmental impacts. The choice of technique may depend on factors such as regional oceanographic conditions, alkalinity source availability, and engineering feasibility. This chapter considers electrochemical methods, the accelerated weathering of limestone, ocean liming, the creation of hydrated carbonates, and the addition of minerals to coastal environments. In each case, the technical aspects of the technologies are considered, and implications for best-practice research are drawn. The environmental and social impacts of OAE will likely depend on the specific technology and the local context in which it is deployed. Therefore, it is essential that the technical feasibility of OAE is undertaken in parallel with, and informed by, wider impact assessments. While OAE shows promise as a potential climate change mitigation strategy, it is essential to acknowledge its limitations and uncertainties. Further research and development are needed to understand the long-term effects, optimize techniques, and address potential unintended consequences. OAE should be viewed as complementary to extensive emission reductions, and its feasibility may be improved if it is operated using energy and supply chains with minimal CO2 emissions.
- Article
(5480 KB) - Full-text XML
- BibTeX
- EndNote




1.1 Overview of ocean-alkalinity-enhancement approaches
The oceans could be artificially alkalized by the addition of alkali (sodium, Na, or potassium, K) and alkaline (magnesium, Mg, or calcium, Ca) silicates, carbonates, oxides, and hydroxides, often as solids, but also in dissolved, aqueous form. The suite of technologies that aim to achieve this is referred to as ocean alkalinity enhancement (OAE). Figure 1 provides a comparative overview of some of the most widely considered OAE approaches.
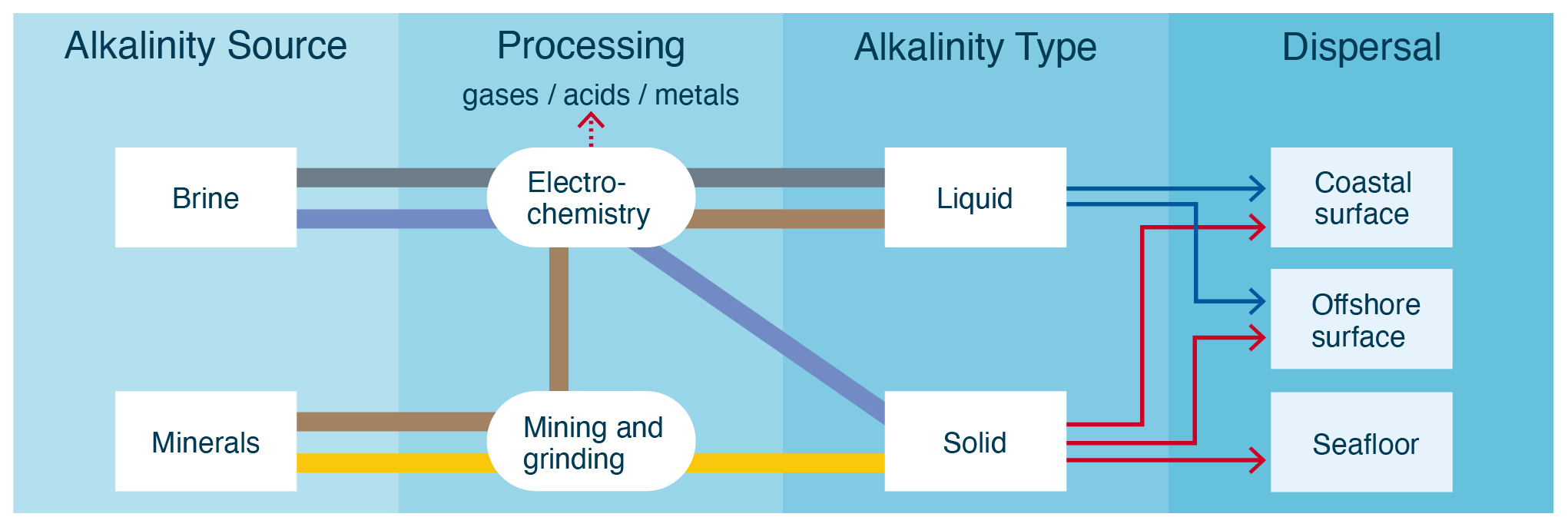
Figure 1Categorization of different OAE approaches by alkalinity source, processing method, alkalinity type, and dispersal location. Each pathway color represents a unique approach. Dispersal location options are determined by alkalinity type. Liquid alkalinity will likely be dispersed on the coast due to its relatively low value of alkalinity per unit volume and mass, whereas solid alkalinity could be dispersed nearshore, offshore, or directly on the seafloor (in shallow water, e.g., <100 m depth).
Approximately 0.25 Gt yr−1 of carbon is removed from the atmosphere due to weathering of silicate rocks on land (Hartmann et al., 2009). The products of this natural process (e.g., Na+, K+, Mg2+, Ca2+, HCO, CO, H4SiO4) are transferred via rivers and groundwaters to the ocean, where they can durably reside for thousands of years (Renforth and Campbell, 2021). This natural process can be enhanced by first mining and grinding the Mg- and Ca-rich silicate rocks such as basalt and peridotite into fine powders and then adding them to the ocean (Rigopoulos et al., 2018; Köhler et al., 2013; Fakhraee et al., 2023). However, for full dissolution of silicate minerals at the ocean surface, very small particles are needed (<10 µm), and as such, the grinding energy to achieve this can be prohibitive (Hangx and Spiers, 2009; Strefler et al., 2018). Instead, larger grains (<100 µm) can be applied to coastal zones (Montserrat et al., 2017), where waves and tides have been suggested to help break down the particles and accelerate dissolution in seawater in close contact with the atmosphere in a process known as coastal enhanced weathering (CEW) (Sects. 6 and 7). Although these larger particles dissolve more slowly, they have a lower initial environmental footprint and could be integrated into coastal management schemes such as beach nourishment (Foteinis et al., 2023).
Carbonate rocks, such as limestone (CaCO3) and dolomite (CaMg(CO3)2), are sometimes proposed as an alternative to silicate rocks for OAE due to their faster dissolution in water. However, the surface ocean waters are supersaturated with respect to calcite and aragonite almost everywhere (Orr et al., 2005), implying that limestone is unlikely to dissolve. One solution is to allow the CaCO3 to sink to deeper water, where it is undersaturated (Harvey, 2008), but the significant delay in making contact with the atmosphere and technical challenges limit this approach. Another solution is to convert the limestone to a more soluble form (i.e., calcium bicarbonate: Ca(HCO3)2(aq)) by first dissolving it in a reactor with a high partial pressure of CO2 (pCO2) (Rau and Caldeira, 1999). This approach is termed accelerated weathering of limestone (AWL) (Sect. 3). Potential improvements to AWL include systems such as buffered AWL, whereby alkaline minerals (e.g., hydrated lime, Ca(OH)2) are added to buffer the unreacted CO2 before being discharged to seawater (Caserini et al., 2021a). Calcium bicarbonate solutions for OAE may also be produced electrochemically (Rau, 2008). For these approaches to be meaningful for CDR (carbon dioxide removal), the concentrated CO2 used in the process must come from the atmosphere (or the surface ocean), via direct air capture (DAC), or from biomass combustion or metabolism.
Alternatively, limestone could be used to create more reactive materials such as lime (CaO) or Ca(OH)2, which dissolve rapidly in the ocean surface – a process referred to as ocean liming (OL) (Kheshgi, 1995) (Sect. 4). Other fast-dissolving solids have been suggested as liming agents, including brucite (Mg(OH)2) (Renforth and Kruger, 2013) and sodium carbonate (“soda ash”, Na2CO3) (Kheshgi, 1995). However, CaO could be mass-produced by the mining, grinding, and then calcining of limestone, potentially using the pre-existing spare capacity of the cement industry (Renforth et al., 2013). The CO2 produced in the calcination step can be stored geologically or even utilized and the CaO, or most likely the Ca(OH)2, transported and spread to the oceans. Nevertheless, open questions remain, particularly around the effect of localized pH increase on the marine ecosystems in the wake of the delivery vessels (Caserini et al., 2021b; He and Tyka, 2023) or pipes, while the potential runaway CaCO3 precipitation could lower the CO2 sequestration efficiency of the approach (Moras et al., 2022).
Alternative pathways are being explored to cost-effectively hydrate minerals and use them as reactive alkaline feedstocks (Sect. 5). Ikaite is one example of a hydrated calcium carbonate mineral which is not supersaturated in the ocean, making it potentially viable for OAE (Renforth et al., 2022). In general, hydration of carbonates has the potential to be less energy-intensive than calcination of limestone while offering comparable alkalinity enhancement to lime and slaked lime.
Aqueous salt solutions such as seawater and brines (e.g., desalination wastes and geological fluids) could potentially provide an abundant source of alkalinity through their electrochemical processing to produce aqueous NaOH(aq) or other hydroxides, which can be used for near-instant OAE and CO2 drawdown (Sect. 2). There are two main methods of electrochemically generating alkalinity from aqueous salt solutions: electrolysis and electrodialysis. Electrolysis (Willauer et al., 2014) produces high-concentration (approximately 26 wt %) NaOH(aq), along with significant quantities of H2(g) and Cl2(g), which must be used within existing energy or product markets or safely stored through reaction with silicate rocks. Electrodialysis (Eisaman et al., 2012) produces lower-concentration NaOH(aq) (approximately 4 wt %) along with HCl(aq) and negligible amounts of H2(g) and O2(g) that are vented. Electrodialysis has a lower theoretical voltage drop than electrolysis per mole of alkalinity generated because it relies on enhancing water dissociation and the subsequent separation of H+ and OH− ions across ion exchange membranes, while electrolysis employs the splitting of water at an electrode surface (Kumar et al., 2021). That said, electrodialysis produces lower concentrations of NaOH(aq) than electrolysis. In electrolysis H2(g) can be burned for energy or utilized, but the Cl2(g) can be difficult to dispose of and is a potential environmental hazard. In electrodialysis, a use or neutralization pathway must be found for the dilute HCl(aq), for example, by neutralization upon contact with abundant sources of mineral alkalinity.
Even though OAE approaches have the potential to remove atmospheric CO2 at the scale of gigatonnes per year, they are also responsible for carbon and other emissions during their life cycle. For example, nitrogen-containing explosives (Tovex) that are typically used for mining can impact eutrophication (Foteinis et al., 2022), whereas nickel (Ni) release from olivine dissolution could contribute to aquatic toxicity (Foteinis et al., 2023), although low solubility or co-precipitation with secondary minerals may limit the impact (Guo et al., 2022). Therefore, for sustainable and scalable OAE the life cycle environmental impacts of each approach should be considered and accounted for via life cycle assessment (LCA), rather than simply relying on carbon balances alone. By doing so, not only is the net carbon dioxide equivalent (CO2 eq.) removal quantified, avoided emissions and tradeoffs with other environmental impacts are also identified. For consistent and meaningful LCAs for OAE, standardized guidelines are required, since the relevant ISO standards (ISO 14040 and 14044) only provide generic guidance that is not technology- or sector-specific. For this reason, the LCA should be specially tailored to OAE applications. Previous work on LCA best practices for similar sectors, such as those for direct air capture and storage (DACS) (Cooney, 2022), and for the wider CDR sector (Terlouw et al., 2021) can serve as a useful starting point.
1.2 OAE technology readiness level
Technology readiness levels (TRLs), developed by NASA in the 1970s, are an estimate of technological maturity. TRLs are based on a scale from 1 to 9, with 9 being the most mature technology (Heder, 2017). Research institutes tend to focus on TRLs 1 to 4, while the private sector focuses on TRLs 7 to 9. Several OAE approaches lie between TRLs 4 to 7, sometimes called the “Valley of Death”, where neither research institutes nor the private sector prioritizes investment.
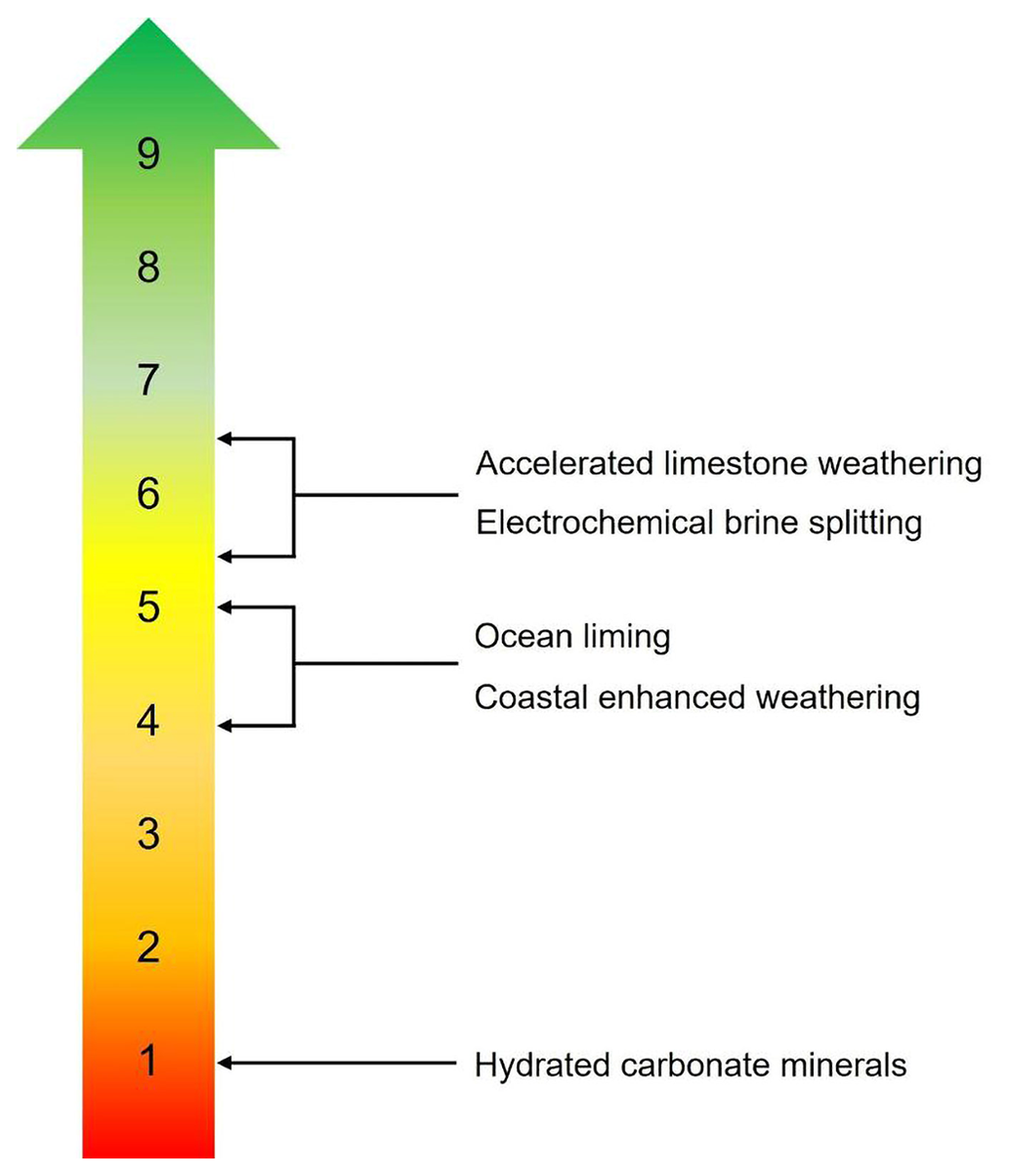
Figure 2Technology readiness levels (TRLs) for different ocean-alkalinity-enhancement approaches: (1) basic principles observed, (2) technology concept formulated, (3) experimental proof of concept, (4) technology validated in laboratory environment, (5) technology validated in a relevant environment, (6) technology demonstrated in a relevant environment, (7) system prototype demonstration in an operational environment, (8) actual system completed and qualified, (9) actual system proven in an operational environment.
The TRL of OAE approaches is summarized in Fig. 2. A feasibility case study for an AWL system attached to a coastal power plant in Taiwan (Chou et al., 2015) and most recently a pilot-scale AWL reactor for flue gas separation (Kirchner et al., 2020b) suggests TRL 5–6. Electrochemical brine splitting has a similar TRL of around 6, with several start-up companies in the process of deploying pilot demonstrations. CEW has a lower TRL of between 4 and 5 and is currently undergoing (at the time of submission) field trials in Southampton, New York, to prove its efficacy. There are still significant challenges to scaling up the approach, particularly surrounding monitoring, reporting, and verification (e.g., Rau et al., 2022; see Ho et al., 2023, this Guide) and potential ecosystem effects (Bach et al., 2019), as well as logistical challenges around mining, grinding, and transporting enough alkaline material from land to distribute in the marine environment, which would require massive infrastructure and long supply chains (Renforth et al., 2013). Previously, OL was assigned a TRL of 3 to 4 (McLaren, 2012) but can now be considered to have advanced to a TRL of 4 to 5 after recent (May 2022) field trials in Florida (Voosen, 2022). Finally, the production and application of hydrated carbonate minerals such as ikaite has a TRL of 1 (Renforth et al., 2022).
Overall, while OAE may have great potential for CDR, there are still many unanswered questions about the long-term ecological impacts and the feasibility of implementing these techniques on a large scale. As such, further research and development is needed to increase the TRLs of these approaches and to determine which, if any, other approaches should be scaled up. The ocean is a heterogeneous system, and field tests are required across a variety of oceanic conditions, e.g., temperatures, upwelling velocities, seawater chemistries, and biological profiles. A research program designed to accelerate technology development and demonstration of pilot-scale facilities will also need to assess any potential ecological impacts and governance issues.
2.1 Technical summary – chloride brines
Aqueous brine (for example, NaCl(aq)) represents an abundant source from which aqueous alkalinity (for example NaOH(aq)) can be generated using electrochemistry. In these approaches, the alkalinity is in the form of hydroxide ions ultimately derived from the water in the brine stream, with the dissolved brine ions (for example Na+ and Cl−) providing the conductivity and charge balance needed for the process.
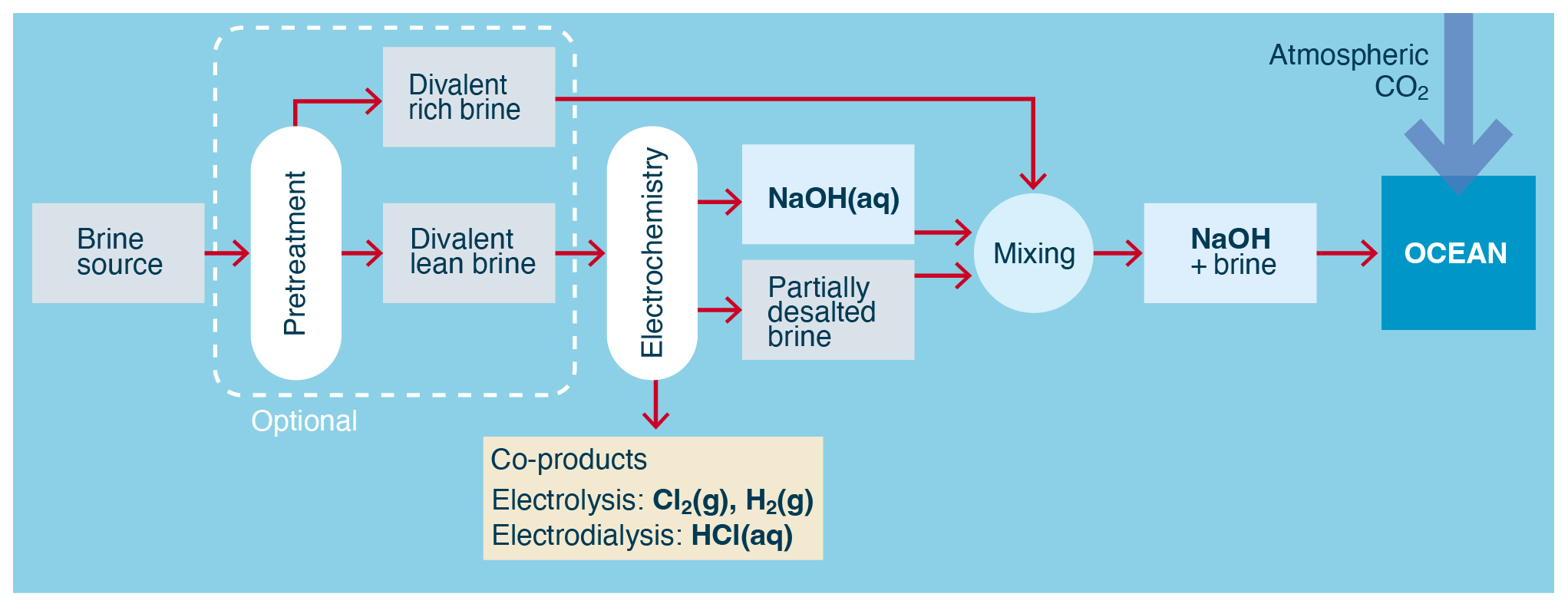
Figure 3Process flow for the electrochemical conversion of aqueous chloride brine into alkalinity. Both pretreatment and the separation of brine into divalent-rich and divalent-lean streams are optional and are not performed in all processes.
The two primary electrochemical processes used to generate alkalinity from brine are electrolysis (O'Brien et al., 2005, pp. 31–34) and electrodialysis (Strathmann, 2011, pp. 163–167), as shown in Fig. 3. Electrolysis generates higher-concentration alkalinity than electrodialysis but requires a higher electrical potential per mole of alkalinity to do so.
This is because electrolysis uses more energy-intensive water splitting at electrodes to generate the alkalinity, while electrodialysis uses enhanced water dissociation at the junction of the bipolar membranes, combined with ion-selective separation. While the primary by-products from electrolytic alkalinity generation are Cl2 and H2 gases, the primary by-product from electrodialytic alkalinity generation is aqueous acid, for example HCl(aq).
The electrolytic generation of alkalinity from an NaCl solution (see Fig. 4) is essentially the well-known chlor-alkali process (O'Brien et al., 2005, pp. 31–34), where aqueous brine (approximately 26 wt %) and NaOH (approximately 28 wt %) are converted into less concentrated brine (approximately 24 wt %), more concentrated NaOH (approximately 30 wt %), hydrogen gas (H2), and chlorine gas (Cl2) (Kumar et al., 2021). The development of efficient and durable oxygen-selective electrodes is critical to making seawater electrolysis more feasible (La Plante et al., 2023).
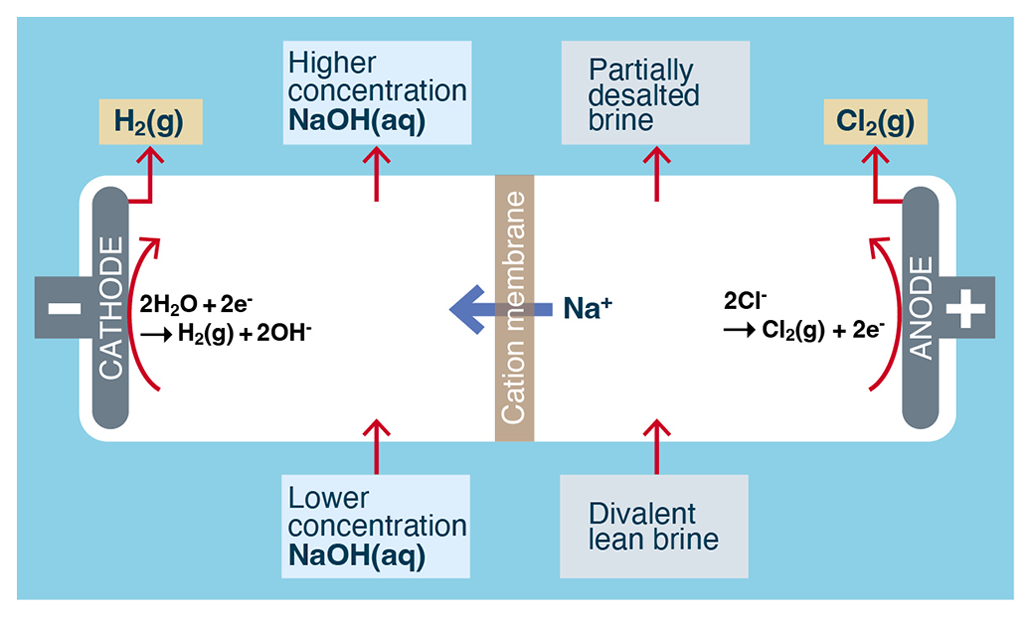
Figure 4Typical process flow for the electrolytic conversion of aqueous NaCl-rich brine into alkalinity using the chlor-alkali membrane process. A chlor-alkali diaphragm process also exists but is not shown (Kumar et al., 2021).
The electrodialytic generation of alkalinity from an NaCl brine typically uses three-chamber bipolar membrane electrodialysis (BPMED) (Strathmann, 2011, pp. 163–167; Fig. 5 herein). In this process, aqueous brine (approximately 3.5 wt %–5 wt %), HCl (approximately 2 wt %), and NaOH (approximately 2 wt %) are converted into less concentrated brine (approximately 2 wt %–3.5 wt %), more concentrated HCl (approximately 3 wt %–4 wt %), and more concentrated NaOH (approximately 3 wt %–4 wt %). Hydrogen gas (H2) and oxygen gas (O2) are created at the end electrodes, but in contrast to electrolysis, because there are typically 50–200 membrane triplets between each pair of electrodes, the rate of H2 and O2 gas generation relative to the rate of NaOH production is negligible, reduced by a factor of the inverse number of cell triplets relative to electrolysis. In practice the H2 and O2 gases generated during electrodialysis are combined and vented. The HCl(aq) generated in this process is used on land in processes that result in the neutralization of the acid, for example in the neutralization of alkaline waste ponds found at sand and gravel operations. Scaling to gigatonnes of CO2 per year will require larger-scale uses of the acid such as the pretreatment of silicate rocks to enhance the kinetics and capacity of CO2 mineralization (Guy and Schott, 1989; Pollyea and Rimstidt, 2017).
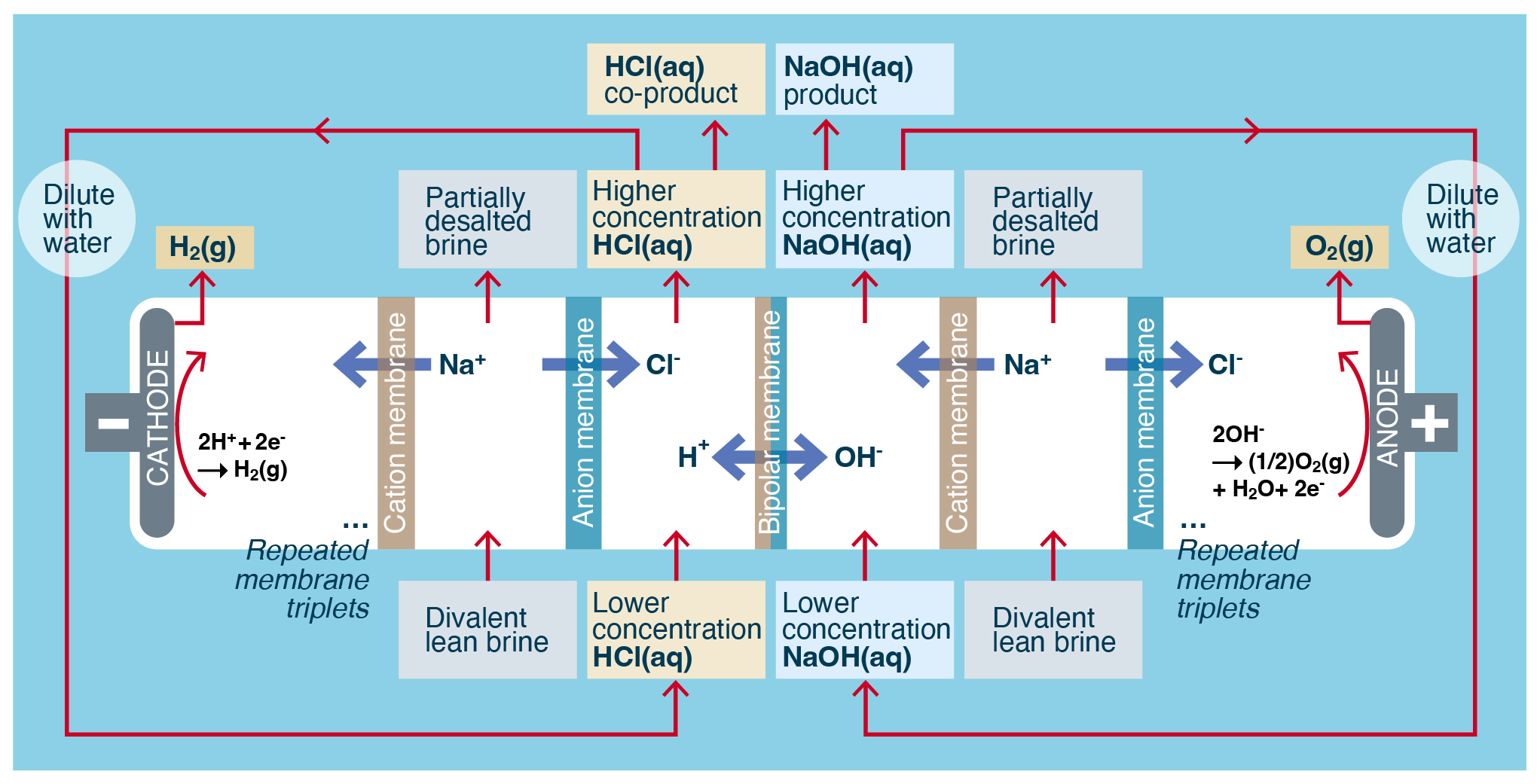
Figure 5Typical process flow for the electrodialytic conversion of aqueous brine into alkalinity.
Once the alkalinity is generated, it must be dispersed into the ocean. An advantage of aqueous hydroxides such as NaOH(aq) is that the rate at which the alkalinity source is added to the ocean is equal to the rate at which alkalinity is actually delivered to the ocean. In contrast, with solid forms of alkalinity such as crushed minerals, the relationship between rate of alkalinity source added and the rate at which this potential alkalinity is delivered to the ocean depends on the dissolution kinetics of the solid. From a molecular point of view, since the Na+ and OH− ions came from the seawater itself, the net effect of the process is simply the removal of acid in the form of H+ and Cl− ions.
Once the alkalinity has been delivered to the ocean, the response of the ocean and atmosphere is governed by two timescales: an immediate timescale that corresponds to the response of the carbonate chemistry of the ocean (Reaction R1),
and a slower timescale of weeks to >100 years (He and Tyka, 2023) that corresponds the re-equilibration of CO2 in the air and surface ocean via air–sea gas exchange (Reaction R2),
The net reaction described by Reactions (R1) and (R2) once equilibrium has been reached is (Reaction R3)
where coefficients a–e are molar ratios relative to the added OH− (added alkalinity), (carbon mass balance), and (charge balance). For example, modeling in CO2SYS (Lewis and Wallace, 1998) shows a=0.827, b=0.742, c=0.086, d=0.086, and e=0.095 in seawater at an equilibrium pH of 8.1, S=35 ppt, T=20 ∘C, and P=1 bar. These ratios are sensitive to the preceding seawater variables; previously reported values for the ratio of moles of removed CO2 to moles of added alkalinity (a coefficients) range from 0.75 to more than 0.85 (Tyka et al., 2022; He and Tyka, 2023; Renforth and Henderson, 2017; Wang et al., 2023; Schulz et al., 2023).
As shown in Reaction (R1), on fast timescales, the addition of alkalinity decreases the dissolved CO2 concentration, putting the surface ocean pCO2 out of equilibrium with atmospheric pCO2. On slower timescales of weeks to months for Reaction (R2), equilibrium is re-established as CO2 from the atmosphere replenishes the CO2 deficit in the surface ocean. The combined result of these two processes is the net removal of CO2 from the atmosphere and storage as oceanic bicarbonate and carbonate ions.
Upon dispersal to the ocean, the added alkalinity is increasingly diluted as it moves away from the point of addition. This results in a mixing zone centered at the point of alkalinity addition where the increase in pH and total alkalinity (TA) is largest. The ratio of the rate of alkalinity addition to the rate of dilution must be kept sufficiently low to avoid the precipitation of Mg(OH)2 (which can result in an undesired increase in turbidity) or CaCO3 (which reduces the efficiency of OAE for CO2 removal) within the mixing zone (Hartmann et al., 2023). Due to this constraint and the permitted limits in the mixing zone for parameters such as pH and turbidity, in practice the pH of aqueous alkalinity may need to be reduced prior to dispersal. For example, prior to release into the ocean, the alkalinity could be mixed with the partially desalted brine stream from which the alkalinity was generated.
To reduce the need for dilution, the alkalinity may first be contacted with CO2 in air to decrease the pH by converting some of the hydroxide (OH−) into carbonate (CO) (Stolaroff et al., 2008). This has the added advantage that all the CO2 captured in this way is measurable and verifiable through direct measurement. This effectively provides a tuneable knob to perform “partial direct air capture” (DAC) to the degree required to reach the target pH, at which point it can be diluted to the pH required by permitting, and the remainder of the CO2 removal can occur via OAE once it is dispersed into the ocean. The advantage of this use of DAC as a “partial pre-equilibration” for OAE compared to standard DAC, is that when used as a preparation step for OAE, no energy needs to be applied to release the CO2 as a pure gas. As an example, if one generates approximately 4 % NaOH(aq) using electrodialysis, partial DAC can be used to bring the pH into the range of 11–12, at which point it can be blended with waste brine to the final pH suitable for ocean delivery.
Rather than partial DAC and dilution, aqueous alkalinity may be directly delivered to the ocean at higher concentrations as long as natural or engineered dilution rates in the mixing zone avoid unwanted precipitation and stay within permitted bounds. Alternatively, when discharged within permitted turbidity limits, more slowly dissolving forms of particulate alkalinity such as Mg(OH)2(s) could be used to distribute the added alkalinity more evenly in space and time (Fakhraee et al., 2023).
A process that is very related to OAE using chloride brines is “indirect ocean capture”, or IOC (de Lannoy et al., 2018; Eisaman et al., 2018; Eisaman, 2020), also referred to as “direct ocean capture”, “direct ocean removal” (DOR), or “ CO2 removal from ocean water” (Kim et al., 2023). This approach employs alkalinity cycling to remove CO2 from the ocean, but without a net increase in ocean alkalinity or DIC (total dissolved inorganic carbon). Because the net alkalinity is not enhanced in this process, it should not be labeled as OAE. In one version of this approach, electrodialysis is first used to generate HCl(aq) and NaOH(aq) from brine streams containing NaCl(aq). The acid is used to acidify seawater, decreasing its pH and alkalinity and shifting all its DIC to dissolved CO2 gas, which is then vacuum-stripped out of the seawater (de Lannoy et al., 2018; Eisaman et al., 2018). The alkaline base is then added to the now decarbonized seawater to restore its lost alkalinity, resulting in CO2 moving from the air to the seawater to restore equilibrium, thereby replacing the vacuum-stripped CO2. At a high level, this approach uses the ocean as a pump, in contrast to OAE, which uses the ocean as a sponge. In a second version of this approach, the NaOH(aq) is added to seawater to remove CO2 as CaCO3(s), with additional NaOH(aq) then added to restore this lost alkalinity and draw CO2 from the air to replace the removed CO2 (de Lannoy et al., 2018; Eisaman et al., 2018). The precipitation of CaCO3(s) reduces alkalinity (resulting either in lower efficiency of CO2 removal per unit of added alkalinity in the case of OAE or a release of CO2 in cases where the precipitation occurs in the absence of an alkalinity addition), making this second version relatively inefficient from a CO2 removal perspective, but this may be pursued if other considerations such as ease of verification outweigh this inefficiency. A third version of this approach relies primarily on the precipitation of Mg(OH)2, in addition to the precipitation of some CaCO3, and prevents release of CO2 in the process of CaCO3 precipitation by generating alkalinity at a sufficiently high rate to keep the pH at a constant target value (La Plante et al., 2021, 2023).
2.2 Technical summary – non-chloride brines and minerals
In addition to the production of alkalinity from chloride salts discussed above, hydroxides can also be electrochemically produced from non-chloride salt solutions such as Na, K, Ca, or Mg sulfates, nitrates, or carbonates. One disadvantage of such an approach is that such salts are much less naturally abundant or less soluble than chloride salts, though they can be present in waste streams. However, as is described in this paper, they can be produced from mineral sources of metal cations and recycled anions. Because of electrochemical issues with nitrate salts and because carbonate salts present more limited net carbonation potential and often have less solubility, the focus here is on metal sulfate salts.
In the case of Na2SO4(aq), such solutions can be electrolyzed or electrodialyzed to produce H2SO4 (sulfuric acid) at the anode and NaOH at the cathode. In electrolysis, H2 and O2 (rather than Cl2) are also produced at the cathode and anode, respectively (Fig. 6).
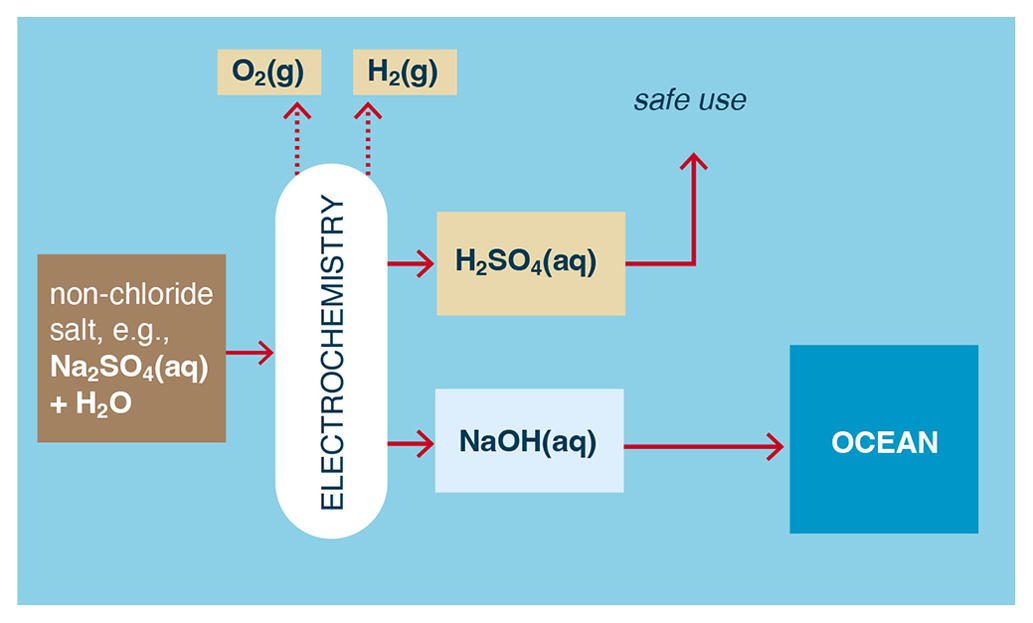
Figure 6Example of a non-chloride metal salt used to electrochemically produce an acid; a metal hydroxide; and, in the case of electrolysis, H2 and O2.
While the NaOH can be used for OAE CDR as described above, uses of the acid at the production scales required for globally significant OAE must be identified. Such acids (including the hydrochloric acid described in the previous section) can be reacted with alkaline minerals to produce more neutral metal salts and water (La Plante et al., 2023). For example, the reaction of sulfuric acid with the silicate mineral forsterite (Mg2SiO4) yields MgSO4, SiO2, and H2O (Reaction R4):
As suggested by House et al. (2007) such metal salts produced from the preceding reaction are in theory benign and could be added to the ocean. However, most silicate minerals contain multiple metals that upon acidification yield metal salts such as Mg, Ca, Fe, Ni, Co, and Na sulfates or chlorides in solution. While at least some of these metal salts will have limited solubility in alkaline seawater, their disposal in the ocean would be problematic due to potential biological effects (see Sect. 4). One alternative is to take advantage of the differences in the reduction in metal solubility as pH rises to selectively remove the less soluble metals as solid metal hydroxides, such as Fe(OH)2, Ni(OH)2, and Co(OH)2, as is commonly done in metal extraction from rocks (Hamilton et al., 2020). Some of the produced NaOH could be used to facilitate pH elevation of the metal salt solution, and the resulting valuable metal precipitates can be harvested for further refining. The remaining, more soluble metal salts, e.g., MgSO4, CaSO4, and Na2SO4, could then provide a more benign way to dispose of the products of acid neutralization.
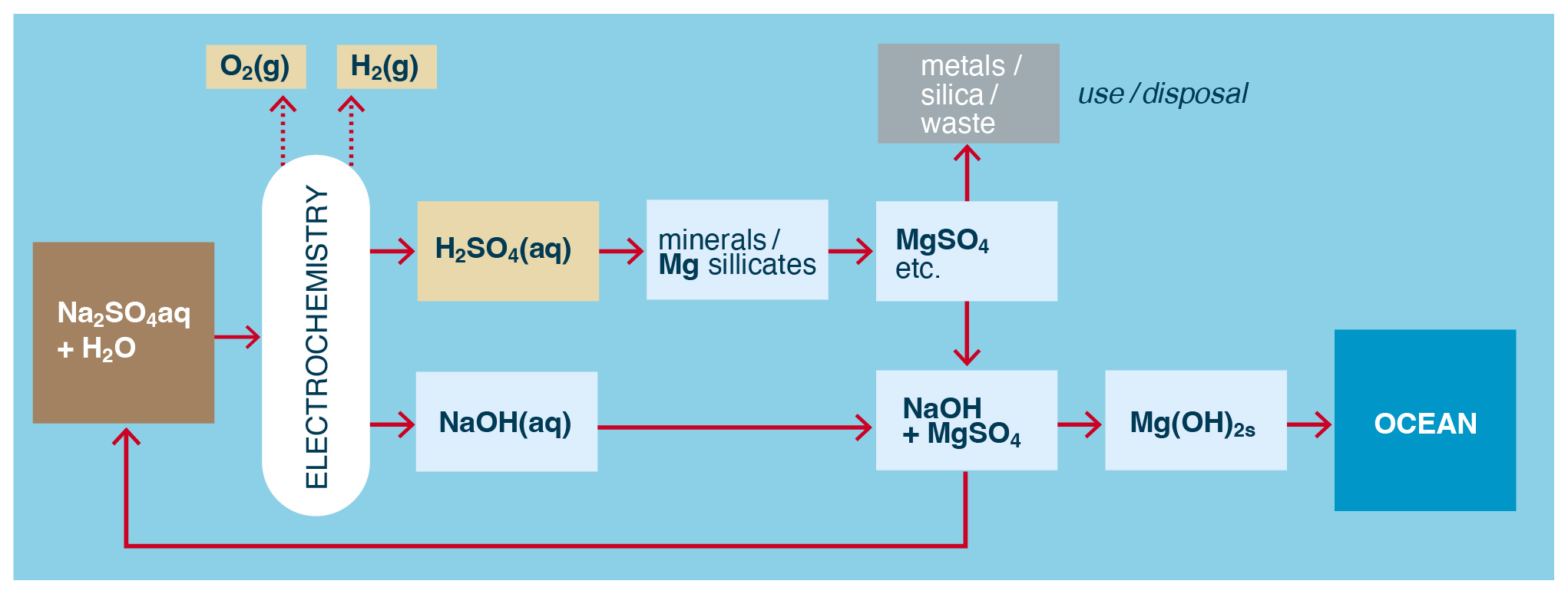
Figure 7Example of the indirect production of solid, polyvalent metal hydroxides from minerals and water using electrodialysis or electrolysis of a monovalent salt. H2 and O2 are also produced.
However, such schemes (e.g., Rau et al., 2013) require loss of SO (or Cl− in the case of HCl use), and thus a continuous supply of (expensive) sulfate would be required. To overcome this challenge, the sulfate can be recycled first as an acid and then as a metal sulfate and back again (Lammers et al., 2023). For example, a monovalent salt solution, e.g., Na2SO4, can be electrolyzed or electrodialyzed to generate H2SO4 that is again used to leach metal salts from minerals, but where the NaOH produced in the catholyte is used exclusively to precipitate less soluble polyvalent metals as metal hydroxides (Fig. 7), with the then-reformed Na2SO4(aq) recycled as brine/electrolyte. In this way Na2SO4 is (largely) conserved, and the metal hydroxide precipitates could then be used as an alkalinity source for OAE if they are at least partially soluble in seawater.
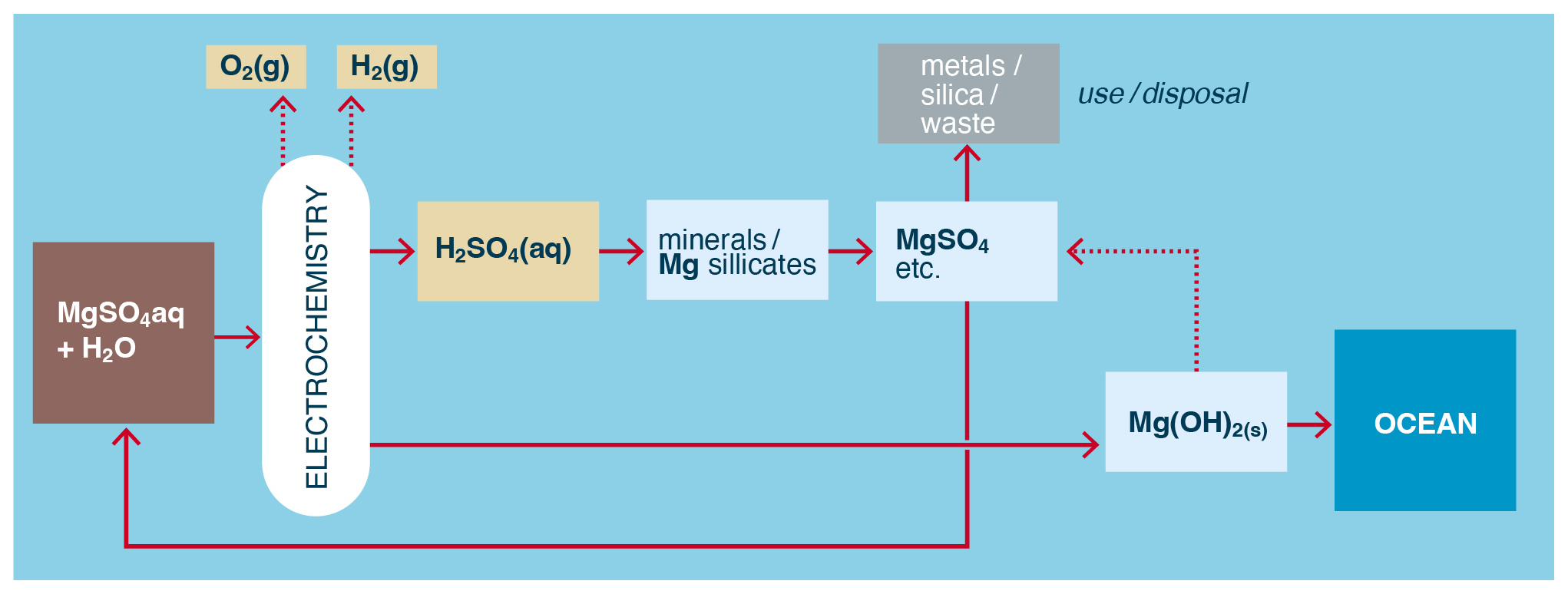
Figure 8Example of the direct use of metal salts acid-leached from minerals in the electrochemical production of alkalinity for OAE.
Another alternative is to use dissolved metal sulfates produced from the mineral/acid leaching directly as electrolyte/brine in cells where the subsequent precipitation of metal hydroxide inside the cell is avoided or otherwise accommodated (Fig. 8). This could include continually harvesting, for example, Mg(OH)2 or Ca(OH)2 precipitated on or near the cathode (Pan et al., 2020; Sano et al., 2018) and/or using diaphragms (Kelland et al., 2022) or membrane-less cells (Talabi et al., 2017) to avoid membrane fouling by precipitates. Compared to the process in Fig. 7, this method more directly generates hydroxides from mineral sources and water.
2.3 Considerations for best research practices
In this section, we highlight key parts of the brine-to-alkalinity OAE process where the application of best practices is especially critical to performing reproducible research.
2.3.1 Brine treatment
The aqueous brine source input into the electrochemical unit for the generation of alkalinity is often seawater, reverse osmosis concentration (ROC), or some other brine stream that also contains the divalent cations Ca2+ and Mg2+. Because the electromechanical systems used for this purpose have compartments with high pH values (>13), care must be taken to avoid the precipitation of solid Mg(OH)2 and/or CaCO3 within the electrochemical unit, as this can lead to increasing voltages, current shunting, and increased hydraulic pressure. Some combination of pretreatment to decrease the concentration of divalent cations and periodic, acidic clean-in-place flushing can avoid this problem. Examples of pretreatment methods include but are not limited to ion exchange and the use of precipitation softening via NaOH(aq) addition.
2.3.2 Alkalinity generation
In practice, the generated alkalinity may have a more complex ionic composition than the simplified description provided above, depending on the input brine composition and the properties of the electrochemical system such as membrane permittivity and selectivity. For example, because seawater contains K+ ions and because cation exchange membranes allow K+ and Na+ transport, the alkalinity generated from the electrodialytic processing of seawater will not be pure NaOH(aq) but will also contain some fraction of KOH(aq). This means that in-line measurement proxies for the generated alkalinity such as conductivity and pH should be calibrated by offline measurements such as TA titrations and inductively coupled mass spectrometry (ICP-MS). Sampling ports built into the electrochemical system are recommended for this purpose.
2.3.3 Aqueous alkalinity dispersal
As previously mentioned, the ratio of the rate of alkalinity addition to the rate of dilution must be kept low to avoid the precipitation of Mg(OH)2 or CaCO3 within the mixing zone. The carbonate chemistry and turbidity should be continuously monitored near the point of dispersal. Precipitation will manifest as a decrease in TA and increase in turbidity (see Sect. 4).
2.3.4 Energy cost, CO2 emissions, and economics
In order for research on electrochemical OAE to be relevant to CDR performed at globally relevant scales, it is necessary to document or estimate the energy use, CO2 emissions, capital costs, and operating costs that are incurred in small-scale systems, and particularly how these would scale for larger systems. These are essential for making informed decisions regarding future RD&D allocations and ultimately decisions about when and where the most cost-effective methods might be deployed.
2.3.5 Environmental and societal impacts and benefits
So as to better inform decision makers, researchers must assess how land, air, ocean, and societal systems might be affected by electrochemical OAE. This includes the environmental and societal consideration of (i) land and resource use such as mineral–salt–brine–water extraction, transportation, processing, and refining; (ii) the footprint of the facility and its operation; and (iii) the downstream impacts/benefits of the products. Researchers should aim to produce a comprehensive budget of all fluxes across the system boundaries (inputs and outputs of energy and matter) to enable this assessment (see Sect. 5).
3.1 Technical summary
In a process called accelerated weathering of limestone (AWL), calcium carbonate (derived from carbonate-bearing rocks, e.g., limestone) can be spontaneously carbonated in the presence of elevated pCO2 and seawater to form predominantly calcium and bicarbonate ions in seawater (Rau and Caldeira, 1999) via the reaction (Reaction R5)
where the molar quantities relative to CaCO3 are approximately a=0.65, b=0.74, c=1.48, d=0.17, and e=0.18 when re-equilibrated with typical seawater at pH 8.1 and at a pCO2=420 µatm. The preceding quantities are halved when expressed in units of moles per mole of alkalinity since 1 mol of CaCO3=2 mol alkalinity. This implies a maximum tonnes of CO2 removal per tonne of CaCO3 of about 0.29 or a minimum requirement of about 3.5 t of CaCO3 per tonne of CO2 captured and stored.
While AWL is an OAE scheme, given the requirement of elevated CO2 to spontaneously drive Reaction (R5), it has been more widely considered to be a CO2 emissions reduction technology, analogous to CCS (carbon capture and storage), at coastally located, fossil-fueled power plants (Rau and Caldeira, 1999; Caldeira and Rau, 2000; Rau et al., 2007; Langer et al., 2009; Rau 2011; Haas et al., 2014; Chou et al., 2015, Kirchner et al., 2020a, b; Caserini et al., 2021a; Xing et al., 2022). However, this approach can be relevant to CDR if the concentrated CO2 used is from (i) emissions from biomass respiration, energy (electricity) production, gasification, or fermentation; (ii) direct air capture; (iii) natural emissions from hydrothermal or geothermal activity; or (iv) possibly natural or artificial upwelling of deep seawater whose pCO2 is high enough and a CaCO3 saturation state low enough to facilitate CaCO3 dissolution.
The CO2 must be of sufficient concentration so that when equilibrated with water or seawater, CaCO3 undersaturation in the solution is effected, and the reaction can spontaneously proceed. Calculations using CO2SYS (Lewis and Wallace, 1998) suggest that surface seawater equilibrated with a pCO2 greater than about 2500 µatm is required for CaCO3 undersaturation to occur and for the reaction to spontaneously proceed. Sufficiently elevated pCO2 drives down solution pH and thus [CO] to achieve a CaCO3 saturation state that is corrosive to CaCO3s. The higher the pCO2, the lower the pH, [CO], and CaCO3 saturation state (Ωcal) and hence the faster the kinetics of the reaction, the greater the areal and volumetric reaction rates achieved, and the higher the DIC concentration attained. Experiments have shown reaction rates ranging from about 10−7 to 10−5 mol m−2 of mineral surface per second. Since volumetric reaction rates are sensitive to carbonate mineral surface area per reaction volume, the interplay among carbonate particle size, seawater and gas contacting, and flow rates dictates reactor design, size, and performance (Rau 2011; Kirchner et al., 2020a; Xing et al., 2022).
Once the calcium bicarbonate+carbonate ions are formed and discharged into the ocean it is presumed that the longevity and security of the storage will be equivalent to that of the existing alkaline C in the ocean, on the order of 100 000 years (Middelburg et al., 2020). This assumes that the AWL Reaction (R5) will not be reversed prematurely by enhanced biotic or abiotic CaCO3 precipitation. Biotic calcification in some marine taxa has been shown to increase with increasing alkalinity and rising calcium carbonate saturation state, Ωcal (Renforth and Henderson 2017; Gore et al., 2019). Note that calcium carbonate saturation state will be more sensitive to the addition of Ca (bi)carbonate than non-Ca alkalinity since both Ca2+ and CO are being added: Ωcal= [Ca2+] [CO]Ksp, where Ksp is a temperature-, salinity-, and pressure-sensitive solubility constant for calcite. Thus, on a per-mole basis, the threshold for carbonate precipitation will be more rapidly reached with calcium-based alkalinity addition relative to the addition of other forms of dissolved metal (bi)carbonates (Fig. 9).
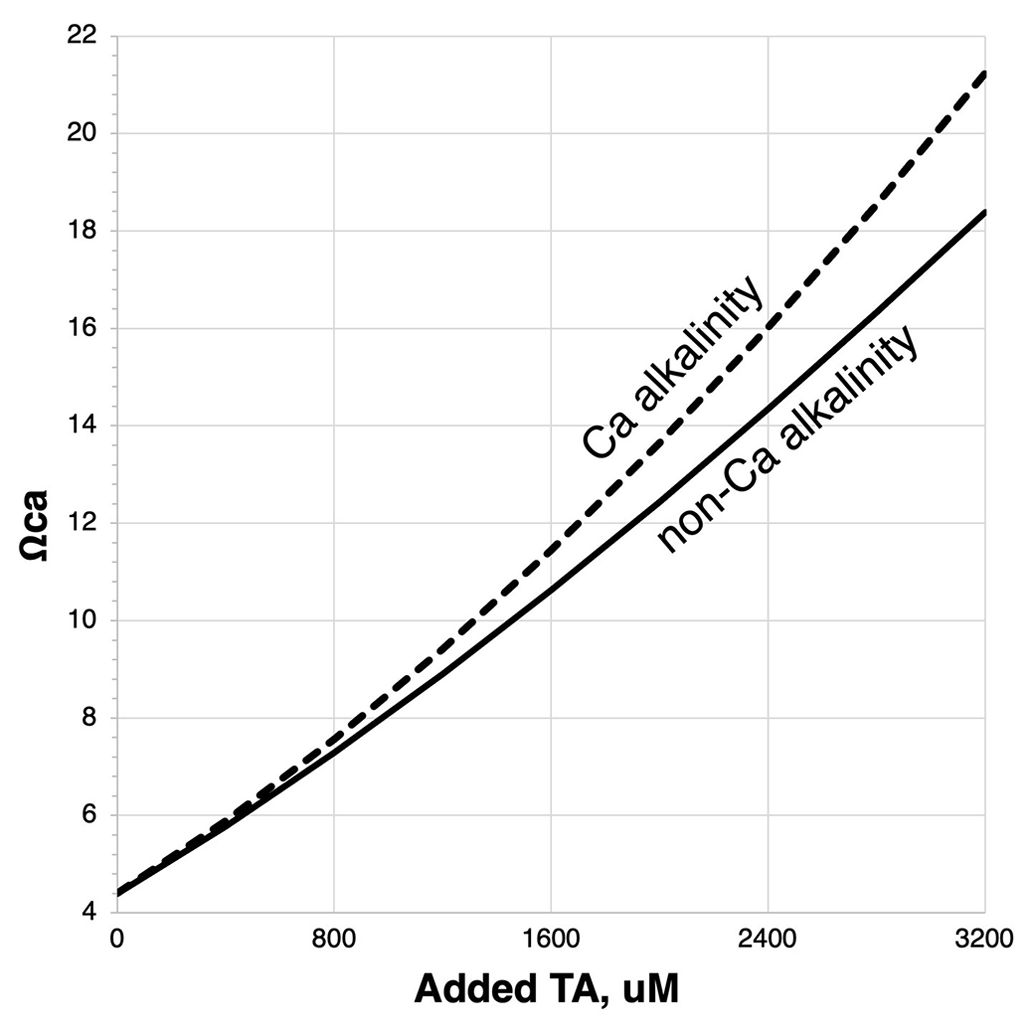
Figure 9Ωca (the saturation state of calcite) response to calcium-based vs. non-calcium-based alkalinity added to seawater initially containing 2350 µM total alkalinity (TA) and equilibrated with a pCO2 of 420 µatm. Modeled using CO2SYS (Lewis and Wallace, 1998) modified to account for variable [Ca2+].
An additional feature that will further promote carbonate precipitation is the degassing of the excess CO2 from the carbonated solution once exposed to air. This in effect removes acid from the carbonated solution, raising pH, [CO], and Ωcal. An example of the chemical sequence of events in carbonating seawater using AWL and then re-equilibrating the carbonated seawater with air is shown in Fig. 10.
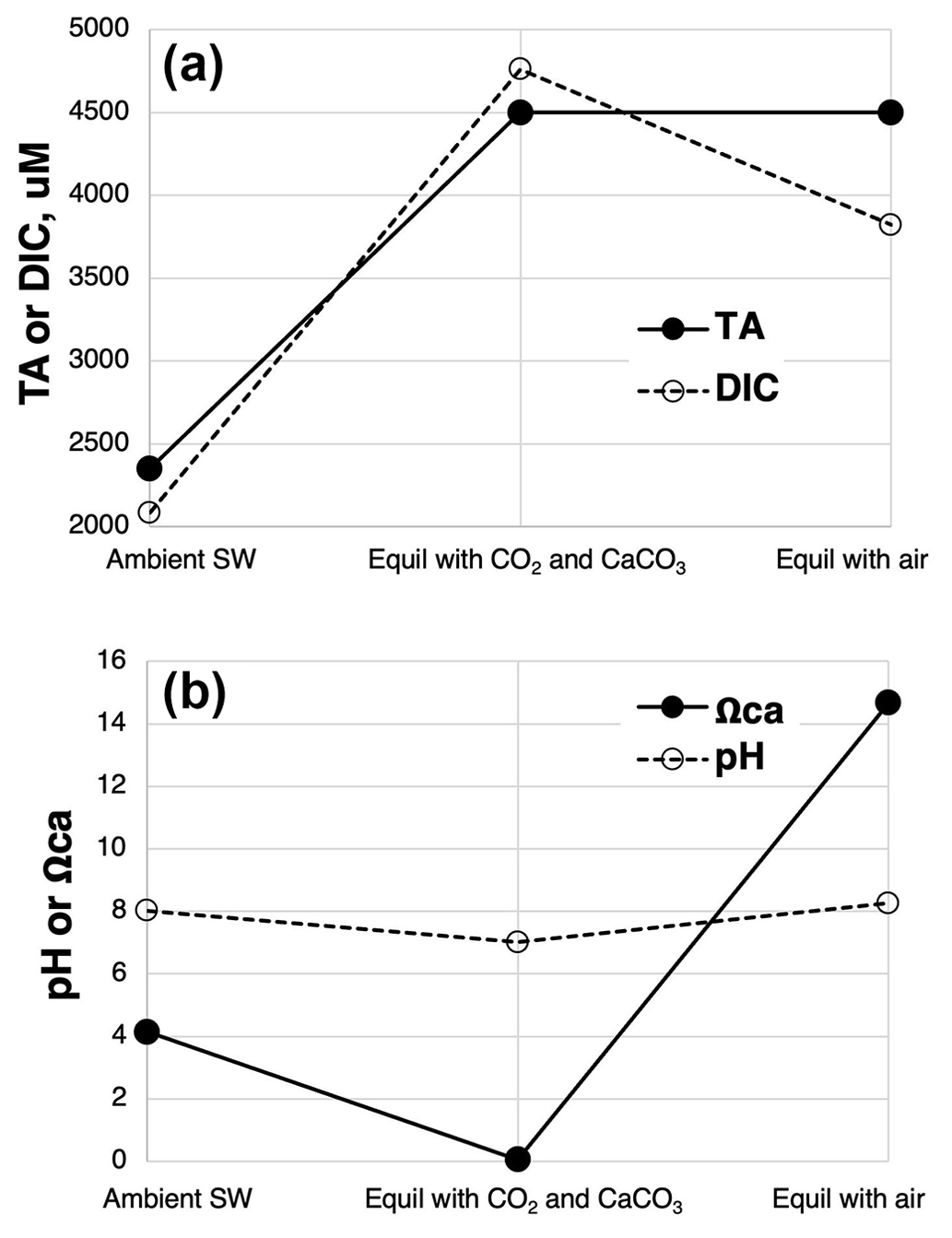
Figure 10Example of the chemical progression of AWL starting first with representative, ambient seawater at pCO2=420 µatm, followed by equilibration with CaCO3s and a pCO2 of 10 000 µatm as well as re-equilibration with air pCO2=420 µatm. (a) DIC: total dissolved inorganic carbon; TA: total alkalinity. (b) Ωca: the saturation state of calcite. Modeled using CO2SYS (Lewis and Wallace, 1998; modified to account for variable [Ca2+]).
Here the net addition of 2150 µM of dissolved calcium (bi)carbonate in equilibrium with an excess of CaCO3s and a pCO2 of 10 000 µtams yields a transfer of carbon from the gas to seawater of 2679 µM, about 310 µM of which is in the form of CO2(aq). If this solution was then added to the surface ocean and allowed to re-equilibrate with air pCO2=420 µatm, pH would rise above that of the initial seawater (by about 0.24 units), and stored carbon would decline from its high value following carbonation by about 35 %, resulting in a net C storage from the AWL process of 1739 µM C (Fig. 10a). In a full-scale facility, Kirchner et al. (2020b) modeled a 50 % loss of captured carbon following an AWL discharge. The percentage of captured CO2 lost will very much depend on the pCO2 of the gas used as well as the degree of chemical equilibration achieved among gas, carbonate, and seawater. Use of high CO2 and its equilibration with seawater, but with incomplete equilibration with carbonate minerals, will result in an AWL discharge solution with high DIC largely as CO2(aq), but with relatively little additional alkaline C formed (little long-term C storage).
Upon equilibration with air, the loss of excess CO2 from the above alkalinized seawater forces a rise in Ωcal to 14.5 in the above example (Fig. 10b). This is >3× higher than the initial ambient seawater and could cause spontaneous CaCO3 precipitation from seawater. However, such an effect is likely countered by the rapid dilution of the carbonated solution with ambient seawater relative to the slow kinetics of CaCO3 precipitation (Fig. 7 in He and Tyka, 2023). As long as dilution occurs faster than CaCO3(s) formation, CaCO3(s) precipitation and alkalinity loss can be avoided (He and Tyka, 2023).
The limitations of this approach in the context of OAE CDR include the need for a concentrated non-fossil CO2 source in close proximity to seawater and carbonate minerals. Potential mineral carbonate sources include globally abundant limestone as well as less abundant dolomite and magnesite. Waste marine shell material or carbonate sands can also be considered, especially because aragonitic shell material should be more soluble than calcite (e.g., limestone) and because this aragonite dissolution simply speeds up the natural return of its marine-derived constituents to seawater. Proximity to the ocean is also a requirement for both the water used for carbonation and for discharge and storage of the carbonated, alkalized seawater. Also, considering a possible upper limit of only about 25 mg C (92 mg CO2) stably stored per liter of seawater, significant pumping of seawater to facilitate gas/carbonate contacting and conversion must occur. Mining, processing, and transportation of >3.5 t CaCO3 t−1 CDR also need to be considered, as do the size and capital and operating costs of the seawater–CO2–carbonate contactor. For one AWL design, Xing et al. (2022) estimate an energy cost of 5.7–8.2 GJ and a land requirement of 7.1–13.1 m2 per tonne of CO2 captured and stored after allowing for degassing of the carbonated seawater following discharge in the ocean.
Advantages of the process include (i) spontaneous, exothermic conversion and long-term storage of CO2; (ii) potential restoration of ocean pH; (iii) relative ease of verifying CDR by carbonating alkalinity prior to release and quantifying the increase in carbon concentration in solution prior to release rather than having to verify CDR occurring in the ocean; and (iv) providing a relatively simple, low-tech, widely applicable approach to OAE at coastal sites.
In sum, considering the global abundance of concentrated CO2 waste streams, calcium carbonate mineral resources (including massive waste piles; Langer et al., 2009), and the reactivity of these minerals in elevated CO2(aq) solutions, AWL seems to be an effective way to perform relatively safe, low-cost, low-tech OAE at scale, especially considering that it is routinely used at small scales to alkalinize saltwater aquaria (Huntington, 2002) and considering that such spontaneous rock–water–CO2 reactions provide the primary source of alkalinity naturally present in the global ocean (Middelburg et al., 2020). Its use in CDR is, however, more restricted considering that the CO2 used must be concentrated above that in air in order to make the CDR rate relevant on human timescales. Further research is needed to better determine the desirability, effectiveness, and capacity of AWL.
3.2 Considerations for best research practices
3.2.1 Purity of feedstocks
Any impurities in the feed CO2 or carbonate mineral have the potential to be released with the discharge of the carbonated seawater. The quantity and impacts of these impurities need to be measured to assess the potential for downstream environmental impact as well as reduction in reactivity per mass of mineral. For example, Kirchner et al. (2020a) found measurable trace metal concentrations in their discharge originating from the limestone used, but all concentrations they considered were “below levels of environmental concern”. Only slightly elevated NO was observed in the discharge seawater that originated from the NOx from the flue gas processed. This study explored the dissolution of limestone in seawater driven by high CO2 from a point source of fossil fuel combustion emissions rather than concentrated from the atmosphere, and it should be noted that these two cases may not have the same impurities.
3.2.2 Monitoring, reporting, and verification
Because AWL carbonation likely occurs before addition to the ocean, the quantity of carbon captured and converted for long-term storage can in theory be easily quantified as an increase in seawater DIC at point of discharge. This is especially true if the carbonated seawater is equilibrated with air prior to release to the ocean so that excess dissolved CO2 (that will ultimately be lost from seawater once released) is not counted as sequestered carbon (Fig. 2). Otherwise, the net CO2 removed and stored can be measured/calculated by either (i) bubbling with air a subsample of the freshly carbonated seawater and measuring its DIC upon air equilibration (e.g., indicated by attaining a stable pH higher than the initially carbonated solution) or (ii) calculating DIC in air-equilibrated discharge by measuring temperature, salinity, pH, DIC, and TA of the freshly carbonated seawater and then modeling its DIC at ambient air pCO2. In each case the proportional difference in DIC before and after air equilibration must be subtracted from the DIC of the carbonated seawater to yield long-term, gross carbon removed. Net carbon removal is obtained by subtracting all anthropogenic CO2 emissions incurred in the performance of AWL from gross CDR: gross CDR − emissions = net CDR. Emissions includes those associated with carbonate extraction, processing and transportation; energy usage in water pumping and other operating activities; and infrastructure and maintenance of the system.
3.2.3 Economics
Estimating the possible economics of AWL systems at scale is essential for making informed decisions on future R&D. It requires extrapolating/modeling the costs measured or inferred at research scales. It is therefore important to record carbonate purity, energy usage, and efficiency as well as volumetric reaction rates, water pumping requirements, etc. so as to better estimate costs and performance at scale.
3.2.4 Environmental and social impacts
As in all OAE, the upstream and downstream environmental and social impacts of AWL must be considered at research and larger scales. In particular the impacts of the following must be considered.
- i.
Increased carbonate mineral extraction and processing. However, it should be kept in mind that limestone mining and processing has resulted in massive waste piles of carbonate material whose use could actually benefit land reclamation (Langer et al., 2009).
- ii.
Seawater pumping, screening, and carbonation, which can impact resident biota (CEC, 2005). When possible, it is best to utilize existing seawater pumping (such as for power plant condenser cooling) to avoid additional cost/impacts of new pumping. If the CO2 used for carbonation is hot (e.g., from exhaust from a biomass energy plant) this will warm the seawater, potentially affecting downstream biota.
- iii.
The purity of the carbonate minerals used. In particular, this refers to the presence of any trace constituents that could have environmental consequences downstream (Sect. 4). Likewise, the purity and temperature of the CO2 used must be considered in evaluating downstream impacts.
- iv.
The societal consequences of AWL activities. This includes those associated with the upstream increased carbonate mineral extraction, processing, and transportation; the footprint of the AWL operation on land; and any impacts occurring downstream in the ocean (see Sect. 5).
4.1 Technical summary
Ocean liming is the process of adding lime (CaO) or hydrated lime (Ca(OH)2) to the surface ocean (Kheshgi, 1995; Renforth et al., 2013), the dissolution of which increases seawater alkalinity (Reactions R6 and R7; Kheshgi, 1995). Lime is conventionally manufactured through the calcination of limestone at >800 ∘C (Reaction R8) using fossil fuels and is used in a range of industries including steelmaking, paper manufacturing, construction, and agriculture.
First proposed for OAE (Kheshgi, 1995), the CO2 produced from the kiln (from limestone decomposition and fossil fuel use) must be captured and stored for the technology to result in a net CO2 removal (Fig. 11). However, energy requirements (<6 GJ per tonne of CO2) and the cost (USD 70–160 per tonne of CO2) of ocean liming are consistent with other engineered CDR approaches (Renforth et al., 2013). Others have shown that the integration of biomass and hydrogen energy vectors may improve the process carbon balance and cost feasibility (Caserini et al., 2019).
Kiln technologies for CaO (often referred to as “burned lime” or “quicklime”) production are diverse and include upright or inclined shafts, rotating shafts, and parallel or contraflow introduction of fuel and feedstock. The selection of kiln type depends on the product material characteristics, quality of the limestone feedstock, local market demand, fuel type and availability, and finance availability (European Commission, 2013).
Limestone decomposes at high temperature by solid-state diffusion of CO2 through the material. The resulting CaO retains the overall volume of the initial calcite but with increased internal porosity (Fischer, 1955). As such it is possible to create lump lime (larger particles of lime produced from similarly sized feedstock limestone).
Powdered Ca(OH)2 is produced by adding a stoichiometric volume of water to CaO (“hydration”; if excess water is used this is referred to as “slaking”; Reaction R9). This hydration reaction is exothermic, resulting in the breakdown of CaO to a fine powder of Ca(OH)2. This is thought to be via a topochemical mechanism (Gartner, 2018), which produces Ca(OH)2 particles around 2–5 µm (Yakymechko et al., 2020); these particles often form larger aggregates of 30–40 µm bound together by weak van der Waals forces (Yakymechko et al., 2020). They are also more porous than CaO, resulting in a higher specific surface area (Moropoulou et al., 2001). There is some literature to suggest that the size of the particle may be controlled by the slaking temperature, (e.g., steam slaking Pesce et al., 2023). Furthermore, the fast reaction rates resulting from small particle size and high surface area are useful in most traditional applications but may not be appropriate for ocean liming (see below):
Provided that the CCS is in place to capture the CO2 emissions from limestone decomposition during calcination, OL can be carbon-negative even when current technology and not fully decarbonized energy is used. Specifically, an LCA on OL (Foteinis et al., 2022) revealed that the main environmental hotspots of the process were limestone calcination, where fossil fuel is consumed for heat generation, followed by CCS, which is energy intensive, while mining, hydration, and ocean spreading affected the environment impact to a much lesser extent. The LCA results were also sensitive to transportation means and distance (although carbonate sedimentary rocks are widely distributed; Fig. 12) and particularly to the kiln technology and fuel type during calcination. When the best available technology is used, along with renewable electricity to drive the process (e.g., calcination using plasma torches), OL's environmental performance is optimized. If the low-grade heat generated during hydration is also recovered and used for district heating, then avoided emissions could also be realized, which can be larger than the process life cycle emissions. In this sense, not only the full amount of CDR is credited to OL, but avoided emissions are also achieved (Foteinis et al., 2022).
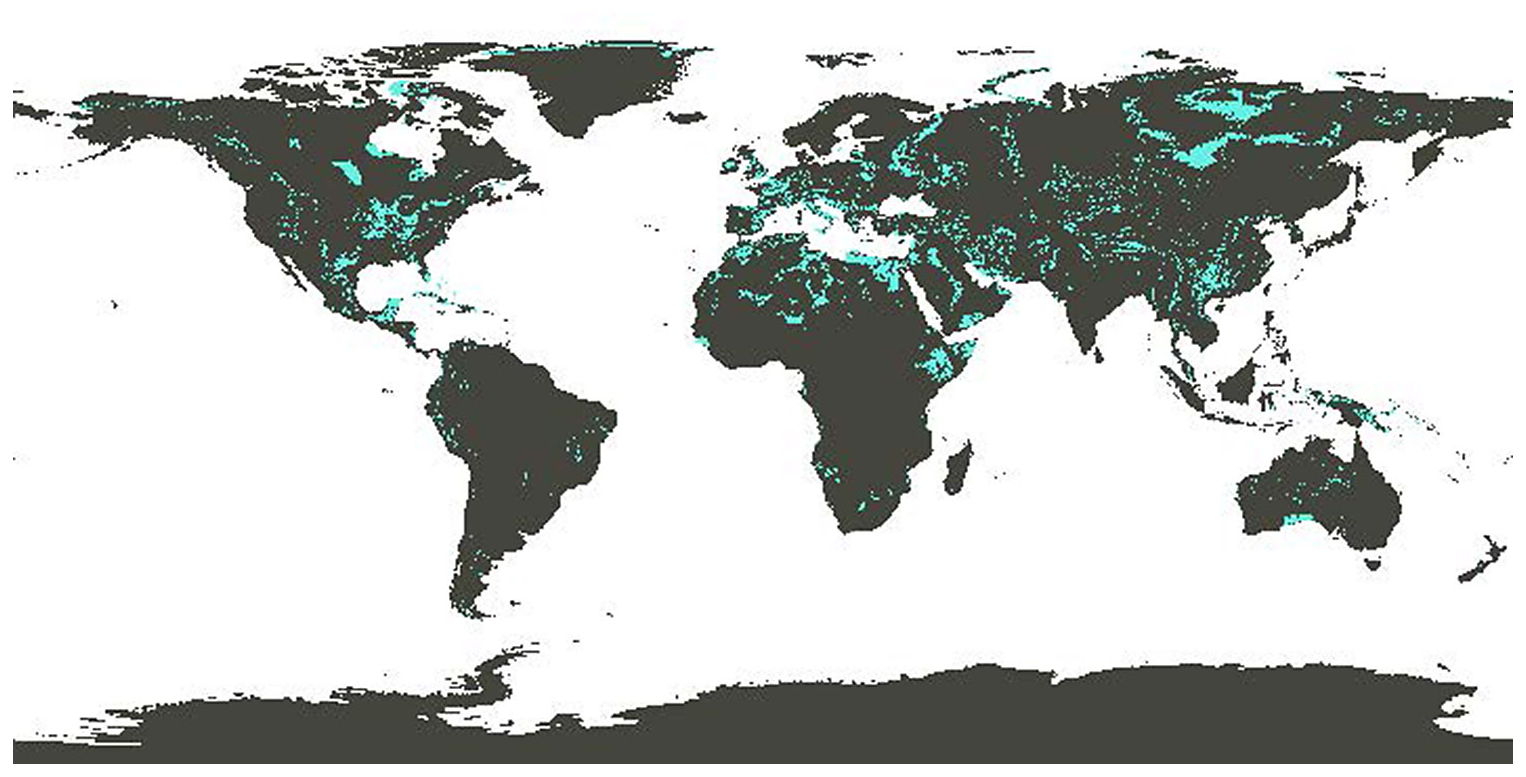
Figure 12Global distribution of carbonate sedimentary deposits (Hartmann and Moosdorf, 2012; map adapted from Renforth et al., 2022).
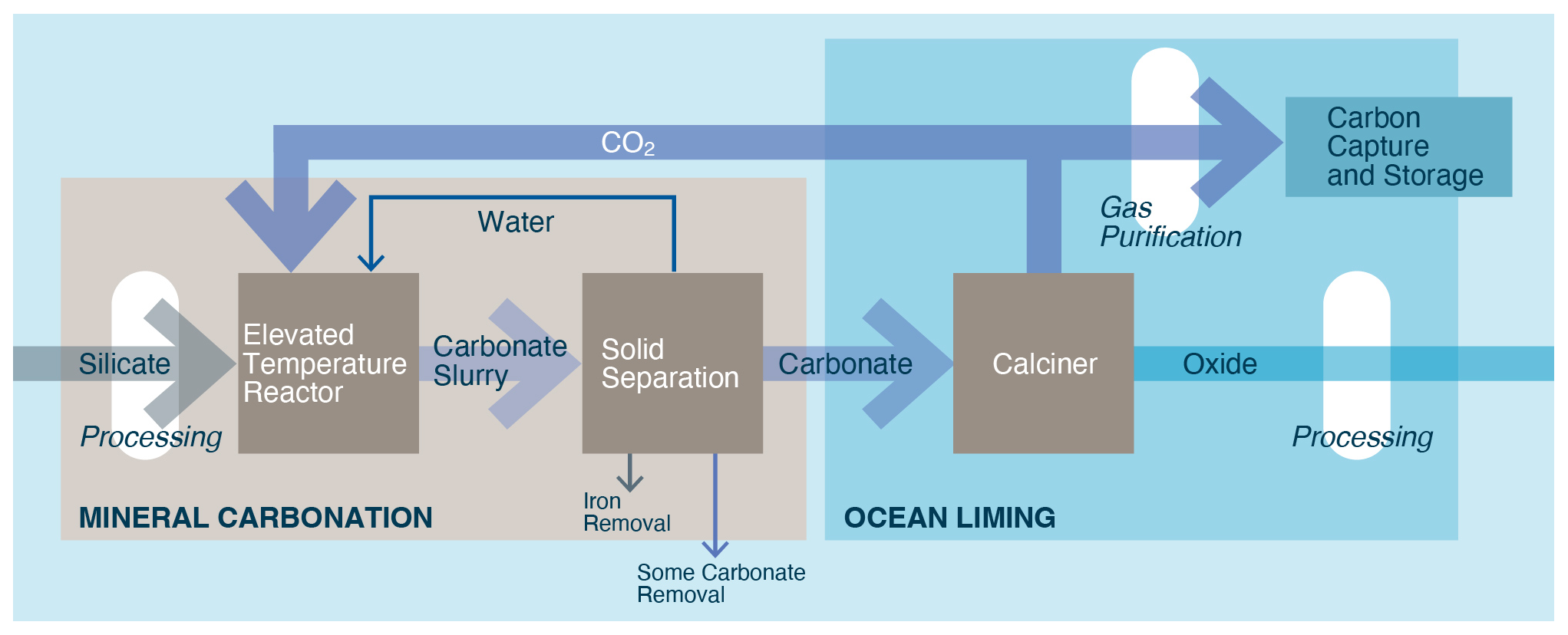
Figure 13A coupled mineral carbonation ocean liming system proposed by Renforth and Kruger (2013).
It is possible to create magnesium oxides and hydroxides, through either the calcination of magnesium carbonates (McQueen et al., 2020) or extraction from magnesium silicates (Renforth and Kruger, 2013). While the calcination temperature and energy of magnesite is substantially lower than that of calcite, global reserves are approximately ∼ 10 Gt (McQueen et al., 2020), which suggests that their exploitation at scale for ocean liming may be limited. Renforth and Kruger explore the coupling of mineral carbonation and ocean liming, in which Mg is extracted from abundant silicate minerals through carbonation and then calcined to produce magnesium oxide/hydroxide materials (Fig. 13).
Processes have been suggested for the extraction of Mg from silicate minerals and the creation of Mg(OH)2 for the purpose of capturing carbon dioxide from flue gases (Madeddu et al., 2015; Nduagu et al., 2012) but may be suitable for OAE as well. Nduagu et al. (2012) suggest a multistep process in which serpentinite (a Mg-rich rock) is heated at 400 ∘C, with solid ammonium sulfate creating a solid magnesium sulfate and silica and evolving ammonia and water as gas. The silica can be leached by washing the solid product with water, after which bringing the ammonia gas back into contact with the Mg sulfate creates high-pH conditions under which Mg(OH)2 can precipitate. An alternative approach (Madeddu et al., 2015) heats solids of NaOH and an olivine-rich rock at 180 ∘C, forming Mg(OH)2 and a Na silicate. Both approaches propose using the Mg(OH)2 for direct reaction with flue gas, and no work has explored their potential for OAE.
4.2 Considerations for best research practices
4.2.1 Lime/hydrated lime production
Lime can be easily created for small-scale laboratory experiments by calcining limestone (or laboratory grade calcium carbonate) in a furnace at 900 ∘C. It is possible to sinter lime at temperatures >1100 ∘C, which would result in a lower reactivity. While sintering is often undesirable for commercial lime, the effect may be useful for OAE to reduce particle dissolution rate and prevent oversaturation of carbonate minerals in seawater. Lime products can also be sourced from commercial suppliers. The reaction of burnt lime (CaO) with water is highly exothermic, and its fire safety risk should be considered when storing or using in the laboratory.
4.2.2 Carbonation prior to experimentation
Lime and hydrated lime readily react with atmospheric CO2 and should thus be produced or sourced as near to the start of the experimental work as possible. The materials can be stored in airtight and/or desiccated containers to minimize carbonation. However, it is difficult (if not impossible) to limit carbonation, and it will certainly be present within commercially sourced material. Carbonate content should be measured (e.g., through mass loss on ignition) before the experiment.
4.2.3 Reactivity and spontaneous precipitation in seawater
Commercially sourced hydrated lime has been manufactured for applications in which high reactivity is desirable. Hartmann et al. (2023) have shown that additions of these particles (0.7 mg Ca(OH)2 : 1 g seawater) may result in spontaneous precipitation of carbonate minerals. Caserini et al. (2021b) modeled an initial particle density of approximately 80 mg Ca(OH)2 : 1 g seawater, diluting to <0.6 mg Ca(OH)2 : 1 g seawater (assuming a 10 kg s−1 addition through a single discharge nozzle in the wake of a ship) within about 30 s. Experiments that add hydrated lime to solution should use an initial concentration <0.7 mg Ca(OH)2 assessing particle dissolution across a 102–104 min range of dilution.
5.1 Technical summary
There are several hydrated carbonate minerals such as ikaite, monohydrocalcite, nesquehonite, hydromagnesite, and amorphous calcium carbonate that are undersaturated in the surface ocean and are thermodynamically likely to dissolve and increase alkalinity when added to seawater (Table 1). The occurrence of these minerals and a method for their industrial production is presented below.
Table 1A summary of potential hydrated carbonate minerals for ocean alkalinity enhancement (see Renforth et al., 2022).
* Negative values denote an exothermic/spontaneous reaction.
Ikaite (CaCO3 • 6H2O) precipitates from aqueous solutions close to freezing conditions (Boch et al., 2015) in a narrow temperature range below ca. 4–8 ∘C and depending on ionic strength down to negative temperature values (e.g., in highly saline solutions down to −8 ∘C; Hu et al., 2014; Papadimitriou et al., 2014). Alternatively, elevated-pressure conditions (>3 kbar at 25 ∘C) facilitate the crystallization of ikaite (Marland, 1975; Shahar et al., 2005). The solubility of ikaite is higher compared to the anhydrous calcium carbonate polymorphs calcite, aragonite, and vaterite (Brečević et al., 1993; Marion, 2001). Dissolved compounds such as magnesium, phosphate, sulfate, and organic molecules that inhibit the formation of anhydrous calcium carbonates favor the nucleation of ikaite (Brooks et al., 1950; Bischoff et al., 1993). Outside these restricted environments ikaite dehydrates and disintegrates rapidly (within minutes to weeks), preferentially into more stable carbonate minerals and water (Mikkelsen et al., 1999). In some cases, calcite pseudomorphs after ikaite might persist (“glendonite”; Greinert and Derkachev, 2004).
Monohydrocalcite (MHC: CaCO3 • H2O) is a rare mineral in geological settings (Nishiyama et al., 2013) but is frequently associated with other calcium and magnesium carbonate minerals, such as calcite, aragonite, lansfordite, and nesquehonite (Nishiyama et al., 2013). Monohydrocalcite has been observed in air conditioning systems (Nishiyama et al., 2013), in “moonmilk” deposits in caves (Nishiyama et al., 2013), and in beach sands formed from algal spume (Nishiyama et al., 2013). It has been reported as a significant component of the decomposition of ikaite in the towers of the Ikka Fjord, West Greenland (Nishiyama et al., 2013). Both laboratory studies and natural observations have indicated that the formation of MHC requires the presence of magnesium in the solution (Nishiyama et al., 2013), possibly forming via a Mg-rich amorphous precursor (Nishiyama et al., 2013).
The magnesium carbonate mineral nesquehonite (MgCO3 • 3H2O) precipitates at room temperature from supersaturated solutions rich in magnesium and bicarbonate (Hopkinson et al., 2008). It is metastable and transforms into hydromagnesite under ambient conditions (e.g., Kazakov et al., 1959), which may be responsible for some naturally occurring hydromagnesite (Davies and Bubela, 1973). Mafic and ultramafic mining wastes, by virtue of their high calcium and magnesium content, are prone to forming numerous carbonate species upon contact with atmospheric CO2 depending on the environmental conditions that prevail at the stockpiles, but metastable nesquehonite was reported to be the dominant magnesium carbonate forming under ambient conditions (Zarandi et al., 2017). Upon rising the temperature above 50 ∘C, nesquehonite evolves into thermodynamically more stable products with lower CO2:Mg ratios (Zarandi et al., 2017).
Hydromagnesite (Mg5(CO3)4(OH)2 • 5H2O) is a naturally occurring hydrated magnesium carbonate (e.g., Königsberger et al., 1999). At the Woodsreef Asbestos Mine, New South Wales, Australia, weathering of ultramafic mine waste sequesters significant amounts of CO2 in hydromagnesite (Mg5(CO3)4(OH)2 • 4H2O) (Oskierski et al., 2021). Mineralization of CO2 in aboveground, sub-aerially stored tailings is driven by the infiltration of rainwater dissolving Mg from bedrock minerals present in the tailings (Oskierski et al., 2021). Complete dissolution of source minerals, or formation of Mg-poor products during weathering, is expected to transfer Mg into solution without significant alteration of the Mg isotopic composition (Oskierski et al., 2021). The main mineral sources of Mg in the tailings (silicate, oxide/hydroxide, and carbonate minerals) are isotopically distinct, and the Mg isotopic composition of fluids and thus of the precipitating hydromagnesite reflects both isotopic composition of source minerals and precipitation of Mg-rich secondary phases (Oskierski et al., 2021). The consistent enrichment and depletion of 26Mg in secondary silicates and carbonates, respectively, underpins the use of the presented hydromagnesite and fluid Mg isotopic compositions as a tracer of Mg sources and pathways during CO2 mineralization in ultramafic rocks (Oskierski et al., 2021).
Amorphous calcium carbonate (ACC) is unstable under normal conditions and is found naturally in taxa as wide-ranging as sea urchins, corals, mollusks, and foraminifera. It is usually found as a monohydrate, holding the chemical formula CaCO3 • H2O; however, it can also exist in a dehydrated state, CaCO3 (Rodriguez-Blanco et al., 2011). ACC was first reported over 100 years ago, when a non-diffraction pattern of calcium carbonate was discovered by Herman Sturcke, exhibiting its poorly ordered nature (Rodriguez-Blanco et al., 2011). The structure and chemistry of ACC is complex, with several forms of ACC classified according to their water content, local order, and mode of formation (e.g., abiotic vs. biogenic). A key variable is the amount of structural water. Hydrated ACC can contain up to ∼1.6 mol of water per mole of CaCO3, yet several less hydrated and even anhydrous forms of ACC have been described (Rodriguez-Blanco et al., 2011; Bots et al., 2012).
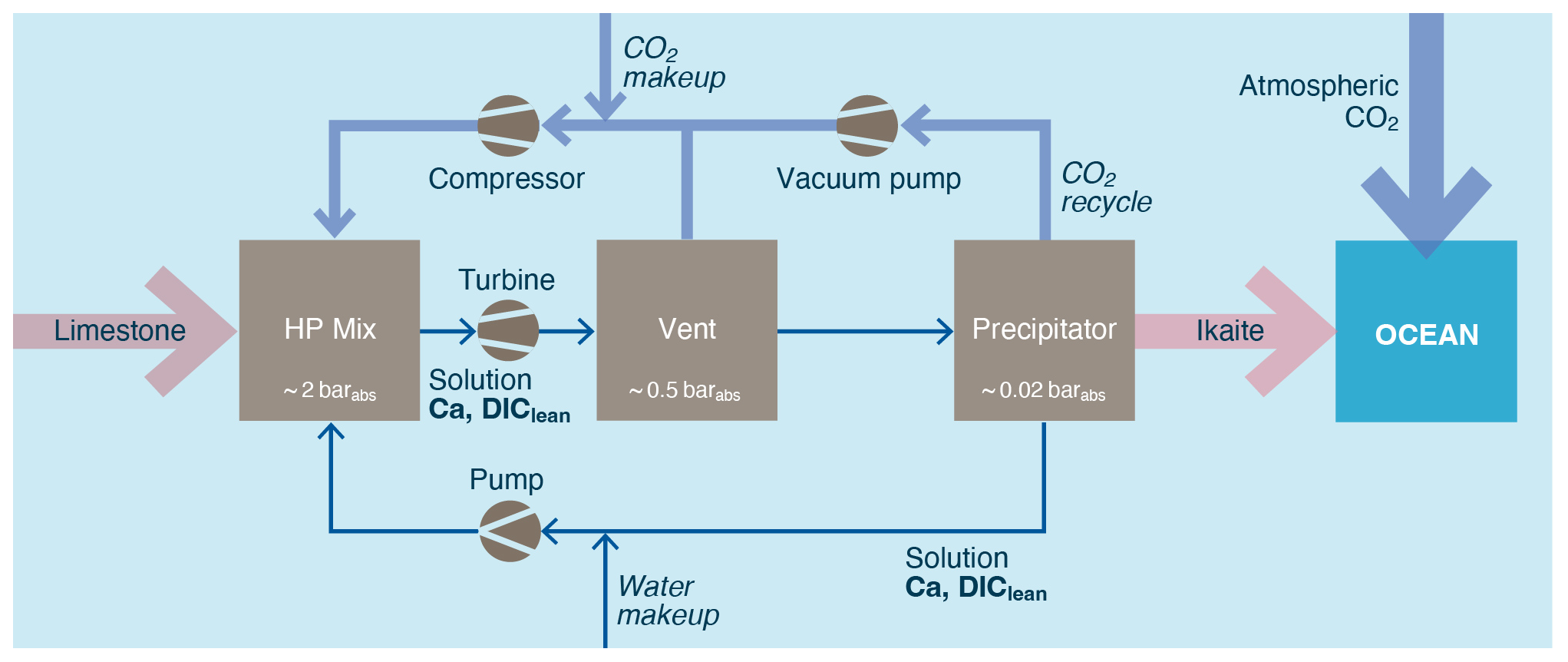
Figure 14A CO2 pressure swing process for creating a hydrated carbonate (adapted from Renforth et al., 2022).
Hydrated carbonate minerals are relatively rare and thus insufficient to meet the demand of a scaled up OAE industry. As such, they would need to be created. Renforth et al. (2022) suggest a process by which limestone is dissolved in water in an elevated CO2 pressure reactor (∼ 2 bar). The water is then degassed in lower-pressure reactors (∼ 20 mbar) under vacuum to evolve/recycle the gaseous CO2 and create conditions under which carbonate minerals are likely to precipitate from the solution (Fig. 14). If the precipitation environment is cooled or in the presence of calcite precipitation inhibitors, then Renforth et al. (2022) suggest that ikaite will form.
There are several benefits for considering hydrated-carbonate-mineral addition for OAE. The chemical energy imparted into lime or hydrated lime during production is released during its dissolution and carbonation in the surface ocean. This energy is impossible to recover. The production of hydrated carbonates requires considerably less energy.
5.2 Considerations for best research practices
5.2.1 Summary of method of precipitating hydrated carbonates
The multistep pressure system for its mass production (Fig. 14) may not be convenient or necessary for all laboratory studies that may want to consider the impact of ikaite dissolution on seawater biogeochemistry. Existing methods for precipitating hydrated carbonates involve the reaction of an alkaline liquid (usually NaOH or Na2CO3) with calcium or magnesium chloride (e.g., Lennie et al., 2004). It is not yet clear if characteristics of these materials differ from those that might be produced from a pressure swing system.
5.2.2 Stability of hydrated carbonates
The feasibility of using hydrated minerals for OAE requires the produced mineral to remain stable for sufficient time to be added to the ocean and dissolve. Similarly, experiments performed using hydrated minerals should have sufficient confidence that the materials added to seawater have not converted to more stable anhydrous polymorphs. If these minerals were to transform into calcite or magnesite before addition it could result in a reduction in alkalinity by seeding additional carbonate precipitation. Experimental work suggests that synthetic ikaite can be stable for days at low temperature and that it increases alkalinity when dissolved in seawater (Renforth et al., 2022).
5.2.3 Methods of detection
Hydrated carbonate minerals are identifiable by several techniques including Raman spectroscopy, Fourier transform infrared (FTIR) spectroscopy, X-ray diffraction (XRD), electron probe X-ray microanalysis (EPMA), and scanning electron microscopy (SEM).
Raman spectroscopy is a non-destructive method that requires little or no sample preparation. Moreover, all the bands related to carbonates in the mid-infrared region have characteristic positions which make it easy to differentiate from other minerals. Raman analyses of carbonates have long been used in mineralogical and geochemical research (see Kim et al., 2021, and references therein).
FTIR is rapid (a few minutes per sample), does not require hazardous chemicals, has a small sample requirement (∼ 1 mg), and produces several distinguishable carbonate bands in its spectrum. Diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) is a form of FTIR with additional advantages compared to transmission FTIR. DRIFTS does not require sample dilution in infrared transparent material, thus reducing sample preparation time; its sample-holding microcells allow for fixed volumes; and the sample is recoverable after analysis. DRIFTS is a method that has been used for identifying and quantifying calcite and dolomite in natural sediments. Few studies have used spectroscopic techniques to quantify carbonate in non-carbonate geological matrices (Bruckman and Wriessnig, 2013; Du et al., 2014; Tatzber et al., 2007).
XRD is the most commonly used tool for identification of major minerals. In addition to qualitative analysis, quantitative XRD is possible because the peak intensities of a given mineral in the diffractogram are proportional to the weight percent of that particular mineral in the sample. However, peak intensities are also a function of the mineral's absorption coefficient, particle size, degree of crystallinity, and the preferred orientation of the sample; this means that compared to qualitative analysis quantitative XRD requires more specialized expertise to produce accurate results.
6.1 Technical summary
Using the ocean's surface waters (which refers to the mixed layer) for mineral addition to increase ocean alkalinity has been the focus of interest of numerous research projects (Renforth and Henderson, 2017), starting with exploring suitable minerals to achieve increased alkalinity while investigating potential side effects on marine life and ecosystems. An obvious problem for the community of researchers involved in OAE has been identifying and generating the particle type and size required to allow for dissolution, classically considered to be smaller than 63 µm (Hangx and Spiers, 2009). However, while this particle size is comparably easy and energy-efficient to produce, residence times in the water column would for most minerals be too short to allow for dissolution, and particles would sink out too rapidly. To avoid mineral loss via sinking, particle sizes of <1µm would be required (Köhler et al., 2013), which are difficult to produce in a climate-neutral manner, and their application might be harmful for humans, leading to respiratory problems (e.g., Doelman et al., 1990). One way to work with bigger grain sizes is to turn to coastal systems, where particles would sink to the seafloor and be transported back into the water column by natural turbulence, allowing for increased dissolution over time.
Classically, silica minerals such as olivine (forsterite or fayalite) and carbonate sedimentary rock such as limestone and dolostone (dolomite) have been suggested to increase alkalinity in an efficient way (Renforth and Henderson, 2017). However, large-scale experiments are still a rarity (see also Riebesell et al., 2023, this Guide and Cyronak et al., 2023, this Guide). Other possible minerals for this purpose are basalt or serpentine. When choosing minerals, an ambition should be to ensure availability near the application location to reduce the carbon footprint, thus limiting the choice of mineral and again emphasizing the advantage of coastal applications as compared to open-ocean applications.
Another concern when applying alkaline minerals is the stability of alkalinity due to possible formation of carbonate phases. This results from the ocean's supersaturation in calcite and aragonite (Sarmiento and Gruber, 2006). If an increase in alkalinity is introduced along with an increase in carbonate ion concentrations, the supersaturation would increase even more, which has been suggested to lead to solid carbonate precipitation (Fuhr et al., 2021; Moras et al., 2022; Hartmann et al., 2023). This, in turn, would decrease surface alkalinity, causing an effect opposite to the desired one, whereby the decrease in alkalinity would depend on the mineral used. One proposed solution to address this challenge is the application of CO2-equilibrated alkaline solutions to minimize the risk of losing alkalinity due to carbonate phase formation.
Open-ocean and water column silicate mineral applications have the potential to increase both the chemical and the biological carbon pump. Here, the biological carbon pump was of interest due to its potential to remove CO2 on a timescale of several thousands of years (Longhurst et al., 1995; Petit et al., 1999; McNeil et al., 2003). The line of reasoning was often based on Earth's historical considerations, with cold periods in Earth history having been related to increased photosynthetic activity by phytoplankton (Kirschvink, 1992; Penman and Rooney, 2019). However, recent studies suggest that mineral additions can, in addition to benefits, also pose risks to marine life, including primary producers (Bach et al., 2019; Guo et al., 2022). This effect is likely related to the increased concentrations of trace metals enriched in the minerals of choice. Earlier dissolution experiments with olivine have shown that an increase in alkalinity of about 100 µmol L−1 led to an increase of ∼3 µmol Ni L−1 due to non-stoichiometric dissolution of the heterogeneous material; this equals approximately 3 times the natural concentration in seawater (Montserrat et al., 2017), within the toxic range for many eukaryotic microalgae (Glass and Dupont, 2017). Other trace metals present in alkaline minerals, including Cu, Cd, Cr, or other heavy metals, might impose further ecotoxicological effects, depending on their concentration in the minerals transferred to the water column or sediment (Simkin and Smith, 1970; Beerling, 2017). Anthropogenic materials including from mining or cement production could also contain a variety of trace metals at concentrations yet to be determined, which might become particularly problematic to organisms of higher trophic levels in which they accumulate (Garai et al., 2021).
It is essential to consider whether the added minerals remain in the water column and impact the growth of the vital primary producers in the food web. Another important consideration is whether adding alkaline minerals impacts the local communities in the treated area and leads to any changes. Moreover, a possible change in the local communities might see the appearance of organisms that release other greenhouse gases, potentially offsetting the sequestration achieved through the treatment.
Careful considerations have to be undertaken in order to ensure safe minimum standards. One important aspect is that field applications cannot exceed environmental quality standards (EQSs; European Commission, 2017). EQSs define the threshold concentration of potentially harmful toxic metals, like nickel and chromium. The impact of minerals enriched in those trace metals on biodiversity and ecosystem function is difficult to study, as minerals are not necessarily homogeneous and contain similar trace metal concentrations. Trace metal concentrations, however, may limit the amount of mineral addition, especially of olivine-rich rocks, that can be deployed in marine habitats (e.g., Flipkens et al., 2021). The potential difficulty arising from introducing toxic compounds would vary strongly depending on the respective ecosystem's tolerance to those compounds. Coastal environments can have limitations, especially with regard to sediments, which already have a (natural) background in trace metals and accumulate trace metals over time (see Sect. 6.2). Furthermore, seawater pH should be kept within a natural seawater range (pH ≤8.2; Pedersen and Hansen, 2003). Even though phytoplankton can be adapted to a wide range of pH, the growth rates of a majority of species can be influenced significantly (Hinga, 2002; Penman and Rooney, 2019). In addition, a pH >9.5 can promote the precipitation of aragonite, as well as brucite together with phosphorus and silicate (Hartmann et al., 2023). On the other hand, depending on the deployed material, specific plankton groups might benefit from, for example, iron additions (Boyd et al., 2000).
To scrutinize the effect of OAE as a tool to mitigate climate change, it is necessary to also investigate the impact of mineral addition, pH, and alkalinity changes on non-CO2 greenhouse gas production. Two important greenhouse gases are methane and nitrous oxide, with warming potentials of approximately 70 and 300 times that of CO2, respectively (Bange, 2006a). While the ocean is a minor source of methane to the atmosphere, it contributes about one-third of nitrous oxide emissions to the atmosphere (Bange, 2006), making it critical to understand any potential impacts of OAE on its formation. N2O is chiefly produced biologically, with the microbes producing it known to be sensitive to changes in pH (Thomsen et al., 1994; Seeländer, 2023); however, so far, the few available studies indicate a reduction in N2O production if pH and alkalinity are increased.
When considering the technical challenges within the pelagic environment for OAE, one of the primary considerations is how to effectively measure the CO2 offsets generated from techniques that utilize the pelagic environment; therefore a standardization of MRV (measurement, reporting, and verification) is a necessity to secure procedures that ensure the accuracy and precision of measurements (see also Ho et al., 2023, this Guide). Within that, a reliable and well-established analysis technique for determining relevant parameters (e.g., alkalinity) is required to ensure the accuracy and comparability of data. Ecosystem diversity in pelagic environments will vary according to the location because of the biological, chemical, and physical parameters due to, for example, ocean currents, temperatures, and wind. Therefore, considerations from many disciplines are essential to explore the complexity.
To carry out coastal pelagic OAE research an understanding of benthic–pelagic coupling is required to assess the impact on benthic systems and ecosystems and to be able to maximize the use of this coupling for OAE. Further, it is critical to assess the local and regional biodiversity by means of meta-omics, flow cytometry, or similar high-resolution methods for microbial life to then understand what thresholds for trace metal additions would introduce toxic effects as defined in Bach et al. (2019) and references therein. For macro-life forms, targeted ecotoxicological assessments are required to avoid damage to, and heavy metal accumulation in, top predators and to establish robust thresholds.
6.2 Considerations for best research practices
Application of particles is challenging (see also Sect. 7.2), as small sizes are required to assure water column dissolution, but larger particles (∼ 63 µm) can be used if the goal is to achieve OAE in both the benthic and pelagic parts of a coastal system, where resuspension of particles can be useful to achieve mixing. However, any particle addition might lead to shadowing, thus impacting photosynthetic organisms in the pelagic and benthic realm, requiring a thorough understanding of both benthic and pelagic primary production to avoid harming the basis of the ecosystem. In addition, mechanical stress can be imposed on benthic organisms by adding particles; data, however, are limited here and will have to be gathered for every system individually.
While those effects will vary largely for each ecosystem, trace metal toxicity can be avoided. Before particles are added, we recommend a thorough analysis of the major elements in the mineral of choice to quantify the effect of mineral addition on the total alkalinity change potentially obtained in the system but also to define the upper limit of additions with respect to trace metal toxicity. For some organisms, including pelagic primary producers, thresholds are available from the literature; however, to understand the impact of trace metal toxicity on complex food webs testing before application should be carried out in mesocosms and benthocosms containing assemblages or subsets of the natural communities present in the ecosystem of choice. At a minimum, key species should be tested individually for their trace metal (and pH) tolerance to avoid damage to biodiversity.
7.1 Technical summary
Continental shelves comprise only about 7 % of the global surface ocean, yet they account for up to 30 % of the oceanic primary production (Gattuso et al., 1998) and between 10 % and 25 % of the present-day CO2 uptake (Regnier et al., 2013). Shelves are also important areas for cation and TA turnover related to detrital mineral dissolution and authigenic carbonate and clay precipitation (e.g., Linke et al., 1994; Jeandel et al., 2011; Jeandel and Oelkers, 2015; Torres et al., 2020). Assuming an ice-free surface area of continental shelf seas of 22×106 km2, natural carbon uptake in coastal waters has been estimated to be −0.19 Pg C yr−1, showing the importance of shelves as a natural global sink of atmospheric CO2 (Laruelle et al., 2014, 2018).
The addition of ground minerals to the shelf seafloor may enhance this uptake even further and as such are candidate locations for OAE. Large-scale mineral application would be logistically convenient, using excavators either directly on the beach or from small vessels or barges in offshore shallow waters. Several companies and initiatives already take advantage of this relatively easy CDR implementation and study the effectiveness of mineral dissolution in the field.
The most obvious advantage of adding particulate minerals to the seafloor compared to the water column is the required grain size. Compared to water column deployment, for which the required grain sizes are <1µm in order to avoid rapid sinking (Hauck et al., 2016), the optimum grain sizes needed for seafloor deployment are proposed to range between 0.2 and 1.4 mm (Schuiling and de Boer, 2010, 2011; Strefler et al., 2018), which is economically attractive. However, these recommendations lack thorough experimental testing, both in the laboratory and in the field.
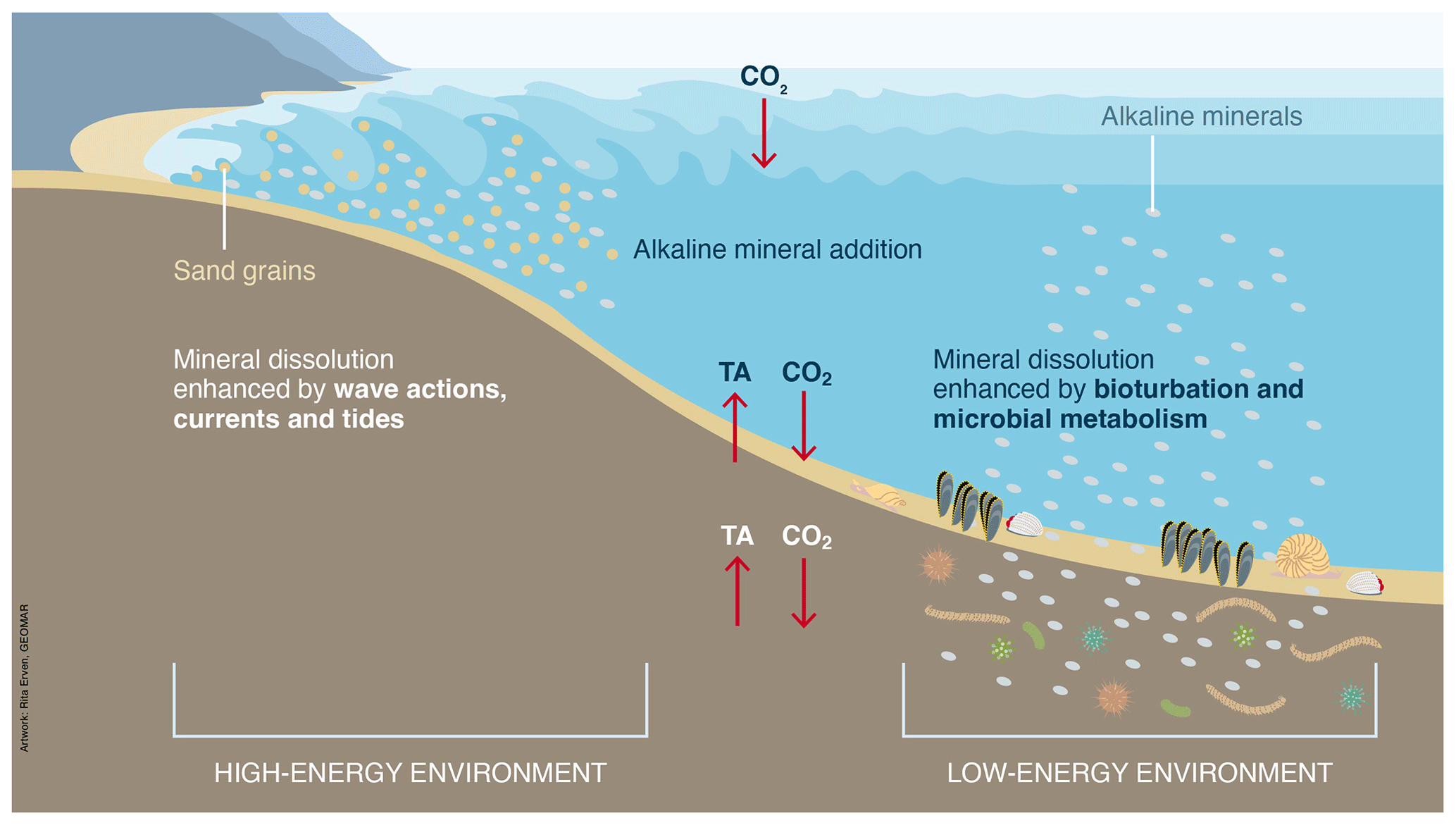
Figure 15High- and low-energy environments with different regional advantages favoring mineral dissolution.
Further, the choice of grain size depends on the region of deployment. Two potential coastal regions for mineral addition are low- and high-energy environments (modified after Meysman and Montserrat, 2017) (Fig. 15). High-energy environments are characterized by extensive water mass movement, such as surf zones, thereby providing a natural grinding mechanism, allowing larger, less costly grain sizes to be used (Flipkens et al., 2023). In contrast, low-energy environments require the addition of smaller grain sizes (∼ 20 to 100 µm) as a higher surface area maximizes mineral dissolution (Oelkers et al., 2018). In this environment, mineral dissolution is enhanced by biota through bioturbation and microbial metabolism (Meysman and Montserrat, 2017). These two distinct environments are discussed below with respect to their advantages and disadvantages.
7.1.1 High-energy environments
These environments are located along the coastal surf zone or the shallow shelf, where wave action, currents, and tidal activity lead to natural mineral erosion. The required grain sizes can be relatively large (millimeter-scale) for this environment, given that the constant movement naturally grinds the minerals, continuously exposing fresh reactive mineral surfaces and enhancing dissolution. Given that alkalinity enhancement directly occurs close to the air–sea interface in these well-mixed shallow water settings, the impact on atmospheric CO2 uptake is immediate.
Table 2Advantages and disadvantages of coastal marine mineral addition on the continental shelf (low-energy environment) compared to the surf zone (high-energy environment).
Detection of mineral dissolution and alkalinity production in these high-energy environments is challenging due to rapid mixing, dilution, and dispersal of solutes. Minerals may also be transported along- and offshore away from their deployment location. This requires carefully designed field experiments to assess the efficiency of this OAE approach with regard to CO2 drawdown (see Sect. 7.2 and Cyronak et al., 2023, this Guide). Furthermore, these factors hamper accurate evaluation concerning MRV for commercial carbon removal purchases (see Ho et al., 2023, this Guide). An advantage of mineral dissolution in coastal surface sediments is the lower risk of secondary mineral formation. In an open environment, supersaturation levels potentially triggering carbonate or phyllosilicate precipitation in conjunction with other relevant factors (e.g., pH, DIC, TA) are more unlikely to be attained. CO2 consumption may therefore be more efficient compared to low-energy environments. See Table 2 for a summary of advantages and disadvantages for mineral deployment in high-energy environments.
7.1.2 Low-energy environments
Shelf environments comprise the waters below the wave base down to 200 m that are not significantly affected by normal wave action. The continental slope and deep seafloor are not considered here given that the exchange of bottom waters with surface ocean waters may be too slow to be relevant for near-term CO2 reduction strategies (Smith et al., 2023).
The shelf sediment is mostly sand and mud (Schulz and Zabel, 2006), providing an ideal environment for mineral dissolution by taking advantage of the “benthic weathering engine” (Meysman and Montserrat, 2017). Here, two mechanisms can be distinguished that potentially accelerate mineral dissolution, that is, microbial metabolism and bioturbation by macro-organisms.
The degradation of organic matter by microbial activity in fine-grained sediment creates a unique microenvironment that may be conducive to dissolution of some types of minerals. The microbial degradation of organic matter by aerobic and, in particular, anaerobic remineralization pathways (e.g., denitrification, iron and manganese reduction, and sulfate reduction) leads to an accumulation of reduced forms of S, N, Fe, and Mn in both the dissolved and particulate phases. If these compounds are exposed to oxygen or nitrate at the sediment surface they can be oxidized rapidly, leading to a local decrease in pH to <6. Under these conditions, carbonate dissolution may take place if pore fluids become undersaturated with respect to the dissolving mineral phase (Jahnke and Jahnke, 2000). Silicate dissolution strongly depends on the mineral type, whereby Mg-silicate dissolution (e.g., olivine) is enhanced at low pH and aluminosilicates at high pH (Oelkers et al., 2018, and references therein). Mineral dissolution may be facilitated or even enhanced through the action of electrogenic cable bacteria that can oxidize S by shuttling electrons from subsurface anaerobic sediments to the oxic surface layer (Meysman et al., 2019). However, cable bacteria are not thought to be highly active in bioturbated sediments, such that their potential impact on alkalinity enhancement by mineral dissolution may not be quantitatively significant at the regional scale.
The effect of burrowing by large macro-organisms, known as bioturbation, enhances mineral incorporation into the sediment matrix and, consequently, brings the minerals in contact with the acidic pore fluids and enhances benthic–pelagic exchange (Neumann et al., 2021). Additionally, the digestive systems of macro-organisms, with their high enzymatic activity, low pH, mechanical abrasion, and digestion, have been shown to increase silicate and carbonate dissolution (Cadée, 1976; Volkenborn et al., 2009). However, this process is poorly understood, and its significance for mineral dissolution is unknown at regional scales.
A large drawback of the low-energy benthic environment for OAE is the high probability of secondary mineral precipitation. Formation of authigenic carbonates or phyllosilicates releases CO2 (e.g., Wallmann et al., 2008; Torres et al., 2020) and directly counteracts the envisioned TA release and CO2 uptake by mineral addition. Authigenic aluminosilicate formation was recently found to be a large Si sink in the global marine Si cycle, releasing CO2 and consuming TA (e.g., Wallmann et al., 2008; Rahman et al., 2017; Tréguer et al., 2021) on timescales of weeks to months, that is, much faster than previously considered (103 years) and impacting element cycles on human timescales (Geilert et al., 2023). Authigenic, inorganic carbonate precipitation at the sediment–water interface or within the sediment column is triggered by alkalinity production from anaerobic microbially mediated reactions (Sun and Turchyn, 2014) and can also be caused by silicate dissolution, buffering the potential pH drop induced by methanogenesis and by that creating carbonate-saturated environments (Torres et al., 2020). In the context of mineral addition to increase ocean alkalinity, these secondary mineral formations may play a major role in the net CO2 sequestration efficiency, as recently shown in laboratory experimental studies (Fuhr et al., 2021; Moras et al., 2022; Hartmann et al., 2023). Further research is required to identify the probability and impact of secondary mineral formation on net CO2 turnover with respect to OAE (Table 2 and Sect. 7.2). The results from laboratory studies though (Fuhr et al., 2021; Moras et al., 2022; Hartmann et al., 2023) are still debatable with regards to their transferability to the open ocean, where secondary mineral saturation states are reached less easily. Mesocosm studies might offer a solution here (Riebesell et al., 2023, this Guide), in which open-ocean conditions can be simulated more realistically, and the triggering factors for secondary mineral formation can be identified (see also Sect. 7.2).
As in the high-energy shallow environments, detection of mineral addition in deeper and fine-grained coastal waters (50–200 m) can be difficult. The deployment of autonomous instruments on the seafloor, such as benthic chambers, can be used to measure fluxes of alkalinity and other dissolved compounds to/from the seafloor. Depending on the nature of the sediment and the carbon degradation rates, benthic chambers can typically detect O2 consumption and nutrient release on timescales of 1–2 d of continuous deployment in shelf environments (Sommer et al., 2016). However, the attribution of mineral dissolution to changes in alkalinity is challenging, particularly against the large seawater alkalinity background. Only in the most reactive coastal settings such as upwelling areas can alkalinity fluxes be determined accurately (Ilyina et al., 2013; Dale et al., 2015). The development and incorporation of high-precision pH and pCO2 sensors may provide a solution to detecting small changes in CO2 and alkalinity fluxes due to mineral dissolution. To our knowledge, the suitability of benthic chambers with respect to detection of OAE at the seafloor still requires field testing.
For both high- and low-energy environments, the risk exists that the alkaline minerals will not remain at the site of deployment, due to seabed erosion by either wave action and currents, transport, or burial. In areas where there are strong bottom currents, fine-grained minerals can be eroded and transported. These minerals are then ultimately delivered to regional depocenters in deeper basins in shallow coastal seas such as the Baltic Sea (e.g., Wallmann et al., 2022) or to the continental slopes on open margins (e.g., Anderson et al., 1994). Once deposited in deep waters, they are removed from the shallow regions, where the benthic–pelagic water mass exchange is rapid.
A final point for consideration is the sedimentation rate. On the one hand, high particulate organic carbon (POC) sedimentation rates are desirable to guarantee high rates of organic matter degradation and low-pH pore fluids necessary for enhanced mineral dissolution. On the other hand, high sedimentation rates of detrital minerals or low POC sedimentation rates may be counterproductive, leading to rapid burial of OAE minerals below the dissolution zone. These factors need to be factored into the cost–benefit analysis of envisaged OAE mineral deployment in low-energy environments.
7.2 Considerations for best research practices
7.2.1 Quantity of deployed mineral
As the effects on the ecosystem by local alkalinity enhancement are still the subject of current investigation, care should be taken when adding minerals to the seafloor. The risk of smothering flora and sessile organisms and clogging of burrows with mineral particles can be minimized by avoiding large deposits in the target area. It is currently unknown whether locally enhanced TA increases in sediment pore fluids driven by oversupply of minerals might be detrimental to certain organisms or produce a shift in the microbial community, potentially affecting mineral dissolution rates. Therefore, dispersed mineral distribution is desirable by, for example, sprinkler systems. Post-deployment channeling of minerals to depocenters by bottom currents might be unavoidable for fine-grained particles. To avoid an accumulation of undissolved minerals on the seafloor, potentially negatively affecting marine ecosystems, care must be taken in assessing the quantity added. An upper limit of long-term mineral addition may be scaled to the local annual POC rain rate depending on the stoichiometry of CO2 sequestration by the relevant mineral if the CO2 released from carbon respiration is quantitatively consumed by benthic weathering. This also assumes that mineral dissolution is tightly coupled to the remineralization of organic matter, which is unlikely to be universally the case. This question is currently being addressed in benthic mesocosm experiments (see Riebesell et al., 2023, this Guide) and still requires verification in field trials. For mafic minerals such as olivine, further unwanted side effects may arise due to release of heavy metals that may be toxic to marine organisms at higher concentrations (e.g., Ni). Testing the potential accumulation of metals in locally sourced sediment cores amended with minerals under laboratory conditions is recommended.
7.2.2 Secondary precipitates
The precipitation of secondary minerals either as discrete grains or on the surface of the added alkaline mineral, decreasing its effective surface area, will hamper the efficiency of CO2 removal. Especially in the case of secondary silicate precipitation, many unknowns remain concerning the controlling factors of formation. Identification of reaction pathways and rates will require highly precise monitoring either of the solid or the fluid phase. Analyses of the fluid phase are challenged by the large background seawater concentrations of chemical tracers, though distinct changes in pore fluid chemistry (e.g., K, Li, Mg) and/or the application of stable-isotope tracers (e.g., Si, B, Li, K) have the potential to identify secondary mineral precipitation. Analyses of the solid phase can identify secondary minerals as well, by either bulk rock quantification techniques (e.g., XRD), in situ mineral analyses (e.g., Campbell et al., 2023), and/or sequential leaching procedures; however, it remains a challenge to identify if the precipitates are of marine or terrestrial origin. For a reliable quantification of secondary mineral formation in the context of OAE, repeated sampling of solid and fluid phases at regular intervals needs to be conducted to assess element turnover and thus CO2 sequestration.
7.2.3 Methods of detection
Arguably the biggest challenge is to quantify and monitor CO2 sequestration related to mineral addition on the seafloor. In low-energy deeper waters, the use of benthic chambers is an option to monitor the carbonate system over discrete time intervals. However, artifacts such as changing redox conditions within the chamber due to oxygen depletion need to be considered and, if possible, compensated for in situ. Regular sampling is also needed. In the shallow and easily accessible high-energy environment, regular water sampling is unlikely to detect alkalinity increase or CO2 drawdown in this highly diluted and well-mixed environment. Long-term monitoring using autonomous chemical sensor platforms for detection of changes in pH, pCO2, and TA (Sonnichsen et al., 2023) is an option but highly challenging due to large fluctuations over timescales ranging from hours to years. Keeping track of the mass of mineral gains over time may yield more robust results.
OAE as a potential solution to combat ocean acidification and remove CO2 from the air continues to show great promise. OAE's unique potential among CDR approaches to compensate for CO2 degassing from the ocean resulting from large-scale atmospheric CO2 removal makes it an especially valuable approach worthy of continued pursuit. This chapter delves into technical aspects of the various technologies and considerations for best practices in their research and development. Although challenges remain, such as cost effectiveness and minimizing environmental impacts, pilot projects have begun to demonstrate the feasibility of deploying various OAE techniques in relevant operational environments. Continued innovation and collaboration among scientists, engineers, and policymakers will be crucial in refining these technologies further. While initial experiments have been successful on a smaller scale, significant challenges lie in deploying these techniques on a global level. Addressing logistical complexities, ensuring proper monitoring and regulation, and securing necessary funding are imperative for successful scaling. Additionally, considering regional variations and selecting appropriate sites for implementation will be vital for maximizing the efficiency and effectiveness of OAE projects. The identification and implementation of best practices are essential for the success of OAE initiatives. Conducting comprehensive environmental impact assessments, employing adaptive management strategies, and promoting transparency and public engagement are crucial steps in ensuring responsible deployment. Learning from past experiences, both positive and negative, will help refine the methodologies and minimize any unintended consequences. It is crucial to approach this strategy with careful consideration, ensuring technology readiness, addressing scaling challenges, and implementing best practices.
Recommendations for research on technical aspects of OAE
-
OAE shows promise as a potential CDR solution, and pilot projects and research activities have begun to demonstrate its feasibility in relevant environments. Continued collaboration among scientists, engineers, and policymakers will be crucial in optimizing these technologies. To maximize the effectiveness of OAE, it is critical to consider location or regionally specific constraints to select appropriate application sites for specific OAE approaches.
-
Conducting comprehensive environmental impact assessments, employing adaptive management strategies, and promoting transparency and public engagement are crucial steps in ensuring responsible deployment of OAE.
-
Excessive accumulation of undissolved minerals on the seafloor, potentially harming marine organisms, should be avoided; assuming that benthic mineral dissolution is coupled to organic matter remineralization, the POC rain rate can be used as an upper limit of the amount of mineral addition.
-
The potential formation of secondary minerals, either carbonates or silicates, which can reduce the net CO2 sequestration efficiency of an OAE approach, needs to be monitored on a regular basis by thorough chemical analyses of the involved fluid and/or solid phases.
-
Technology readiness levels for the different OAE measures need to be increased in laboratory experiments and in field studies, also to identify potential “tipping points” for organism viability and the ecosystem in general.
| AWL | Accelerated weathering of limestone |
| CDR | Carbon dioxide removal |
| CCS | Carbon capture and storage, specifically where CO2 is concentrated from waste streams |
| DIC | Total dissolved inorganic carbon |
| LCA | Life cycle analysis |
| MRV | Monitoring, reporting, and verification |
| OAE | Ocean alkalinity enhancement |
| Ωcal | Calcium carbonate (calcite) saturation state |
| pCO2 | Partial pressure of CO2g |
| pH | Negative logarithm of the hydrogen ion activity, a measure of acidity |
| TA | Total alkalinity |
| XRD | X-ray diffraction |
No data sets were used in this article.
MDE, SG, and PR scoped and edited the contents of the chapter. MDE contributed to Sect. 2, SG contributed to Sect. 7, PR contributed to Sects. 4 and 5, LB contributed to Sect. 5, JC contributed to the Introduction and the Conclusions, AWD contributed to Sect. 7, SF contributed to the Introduction and Sect. 4, PG contributed to Sect. 7, OH contributed to Sect. 4, CRL contributed to Sect. 6, GHR contributed to Sects. 2 and 3, and JR contributed to Sect. 6.
Competing interests are declared in a summary for the entire volume at: https://sp.copernicus.org/articles/sp-oae2023-ci-summary.zip.
Publisher's note: Copernicus Publications remains neutral with regard to jurisdictional claims made in the text, published maps, institutional affiliations, or any other geographical representation in this paper. While Copernicus Publications makes every effort to include appropriate place names, the final responsibility lies with the authors.
We thank the Ocean Acidification and other ocean Changes – Impacts and Solutions (OACIS), an initiative of the Prince Albert II of Monaco Foundation, for its support throughout the project. We extend our gratitude to the Villefranche Oceanographic Laboratory for supporting the meeting of the lead authors in January 2023. Rita Erven (GEOMAR) is thanked for her help in preparing the figures for this paper.
This research has been supported by the ClimateWorks Foundation (grant no. 22-0296) and the Prince Albert II of Monaco Foundation. This research has also been supported by the Horizon 2020 (OceanNETs); the ClimateWorks Foundation (Novel Materials for OAE); the Grantham Foundation for the Protection of the Environment (Ocean Alkalinity Enhancement); and the Bundesministerium für Bildung, Wissenschaft, Forschung und Technologie (RETAKE, DAM mission).
This paper was edited by Lennart Bach and reviewed by Justin Ries and one anonymous referee.
Anderson, R. F., Rowe, G. T., Kemp, P. F., Trumbores, S., and Biscaye, P. E.: Carbon budget for the mid-slope depocenter of the Middle Atlantic Bight, Deep-Sea Res. Pt. II, 41, 669–703, https://doi.org/10.1016/0967-0645(94)90040-X, 1994.
Bach, L. T., Gill, S. J., Rickaby, R. E. M., Gore, S., and Renforth, P.: CO2 Removal With Enhanced Weathering and Ocean Alkalinity Enhancement: Potential Risks and Co-benefits for Marine Pelagic Ecosystems, Front. Clim., 1, 7, https://doi.org/10.3389/fclim.2019.00007, 2019.
Bange, H. W.: New Directions: The importance of oceanic nitrous oxide emissions, Atmos. Environ., 40, 198–199, https://doi.org/10.1016/j.atmosenv.2005.09.030, 2006a.
Bange, H. W.: Nitrous oxide and methane in European coastal waters, Estuarine, Coastal and Shelf Science, 70, 361–374, https://doi.org/10.1016/j.ecss.2006.05.042, 2006b.
Beerling, D. J.: Enhanced rock weathering: biological climate change mitigation with co-benefits for food security?, Biol. Lett., 13, 20170149, https://doi.org/10.1098/rsbl.2017.0149, 2017.
Bischoff, J. L., Fitzpatrick, J. A., and Rosenbauer, R. J.: The Solubility and Stabilization of Ikaite (CaCO3 • 6H 2O) from 0∘ to 25 ∘C: Environmental and Paleoclimatic Implications for Thinolite Tufa, J. Geol., 101, 21–33, https://doi.org/10.1086/648194, 1993.
Boch, R., Dietzel, M., Reichl, P., Leis, A., Baldermann, A., Mittermayr, F., and Pölt, P.: Rapid ikaite (CaCO3•6H2O) crystallization in a man-made river bed: Hydrogeochemical monitoring of a rarely documented mineral formation, Appl. Geochem., 63, 366–379, https://doi.org/10.1016/j.apgeochem.2015.10.003, 2015.
Bots, P., Benning, L. G., Rodriguez-Blanco, J.-D., Roncal-Herrero, T., and Shaw, S.: Mechanistic Insights into the Crystallization of Amorphous Calcium Carbonate (ACC), Cryst. Growth Des., 12, 3806–3814, https://doi.org/10.1021/cg300676b, 2012.
Boyd, P. W., Watson, A. J., Law, C. S., Abraham, E. R., Trull, T., Murdoch, R., Bakker, D. C. E., Bowie, A. R., Buesseler, K. O., Chang, H., Charette, M., Croot, P., Downing, K., Frew, R., Gall, M., Hadfield, M., Hall, J., Harvey, M., Jameson, G., LaRoche, J., Liddicoat, M., Ling, R., Maldonado, M. T., McKay, R. M., Nodder, S., Pickmere, S., Pridmore, R., Rintoul, S., Safi, K., Sutton, P., Strzepek, R., Tanneberger, K., Turner, S., Waite, A., and Zeldis, J.: A mesoscale phytoplankton bloom in the polar Southern Ocean stimulated by iron fertilization, Nature, 407, 695–702, https://doi.org/10.1038/35037500, 2000.
Brečević, L., Nielsen, A. E., Oskarsson, Å., Hewitt, G. M., Kutney, J. P., Li, K., Milanova, R. K., Nakata, H., Nasiri, A., and Okada, Y.: Solubility of calcium carbonate hexahydrate, Acta Chem. Scand., 47, 668–673, https://doi.org/10.3891/acta.chem.scand.47-0668, 1993.
Brooks, R., Clark, L. M., and Thurston, E. F.: Calcium carbonate and its hydrates, Philos. T. Roy. Soc. A, 243, 145–167, 1950.
Brooks, R., Clark, L. M., Thurston, E. F., and Freeth, F. A.: Calcium carbonate and its hydrates, Philos. T. Roy. Soc. A, 243, 145–167, https://doi.org/10.1098/rsta.1950.0016, 1997.
Bruckman, V. J. and Wriessnig, K.: Improved soil carbonate determination by FT-IR and X-ray analysis, Environ. Chem. Lett., 11, 65–70, https://doi.org/10.1007/s10311-012-0380-4, 2013.
Cadée, G. C.: Sediment reworking by arenicola marina on tidal flats in the Dutch Wadden Sea, J. Sea Res., 10, 440–460, https://doi.org/10.1016/0077-7579(76)90020-X, 1976.
Caldeira, K. and Rau, G. H.: Accelerating carbonate dissolution to sequester carbon dioxide in the ocean: Geochemical implications, Geophys. Res. Lett., 27, 225–228, https://doi.org/10.1029/1999GL002364, 2000.
Campbell, J. S., Bastianini, L., Buckman, J., Bullock, L. A., Foteinis, S., Furey, V., Hamilton, J., Harrington, K., Hawrot, O. K., Holdship, P., Knapp, W. J., Maesano, C. N., Mayes, W. M., Pogge von Strandmann, P. A. E., Reershemius, T., Rosair, G. M., Sturgeon, F., Turvey, C., Wilson, S., and Renforth, P.: Measurements in Geochemical Carbon Dioxide Removal, 1st edn., Heriot-Watt University, https://doi.org/10.17861/2ge7-re08, 2023.
Caserini, S., Barreto, B., Lanfredi, C., Cappello, G., Ross Morrey, D., and Grosso, M.: Affordable CO2 negative emission throughhydrogen from biomass, ocean liming, and CO2storage, Mitig. Adapt. Strateg. Glob. Change, 24, 1231–1248, https://doi.org/10.1007/s11027-018-9835-7, 2019.
Caserini, S., Cappello, G., Righi, D., Raos, G., Campo, F., De Marco, S., Renforth, P., Varliero, S., and Grosso, M.: Buffered accelerated weathering of limestone for storing CO2: Chemical background, Int. J. Greenh. Gas Con., 112, 103517, https://doi.org/10.1016/j.ijggc.2021.103517, 2021a.
Caserini, S., Pagano, D., Campo, F., Abbà, A., De Marco, S., Righi, D., Renforth, P., and Grosso, M.: Potential of Maritime Transport for Ocean Liming and Atmospheric CO2 Removal, Front. Clim., 3, 575900, https://doi.org/10.3389/fclim.2021.575900, 2021b.
California Energy Commission (CEC): Issues and environmental impacts associated with once-through cooling at California’s coastal power plants, Report No. CEC-700-2005-013, Sacramento, CA, https://www.waterboards.ca.gov/rwqcb9/water_issues/programs/npdes/southbay_power_plant/docs/updates_022410/2005_Issues_OTC_Energy_Commission_Report_2005[2].pdf (last access: 17 November 2023), 2005.
Chou, W.-C., Gong, G.-C., Hsieh, P.-S., Chang, M.-H., Chen, H.-Y., Yang, C.-Y., and Syu, R.-W.: Potential impacts of effluent from accelerated weathering of limestone on seawater carbon chemistry: A case study for the Hoping power plant in northeastern Taiwan, Mar. Chem., 168, 27–36, https://doi.org/10.1016/j.marchem.2014.10.008, 2015.
Committee on A Research Strategy for Ocean-based Carbon Dioxide Removal and Sequestration, Ocean Studies Board, Division on Earth and Life Studies, and National Academies of Sciences, Engineering, and Medicine: A Research Strategy for Ocean-based Carbon Dioxide Removal and Sequestration, National Academies Press, Washington, D.C., https://doi.org/10.17226/26278, 2022.
Cooney, G.: Best Practices for Life Cycle Assessment (LCA) of Direct Air Capture with Storage (DACS), U.S. Department of Energy, Office of Fossil Energy and Carbon Management, https://www.energy.gov/fecm/best-practices-LCA-DACS (last access: 6 November 2023), 2022.
Cyronak, T., Albright, R., and Bach, L. T.: Field experiments in ocean alkalinity enhancement research, in: Guide to Best Practices in Ocean Alkalinity Enhancement Research, edited by: Oschlies, A., Stevenson, A., Bach, L. T., Fennel, K., Rickaby, R. E. M., Satterfield, T., Webb, R., and Gattuso, J.-P., Copernicus Publications, State Planet, 2-oae2023, 7, https://doi.org/10.5194/sp-2-oae2023-7-2023, 2023.
Dale, A. W., Sommer, S., Lomnitz, U., Montes, I., Treude, T., Liebetrau, V., Gier, J., Hensen, C., Dengler, M., Stolpovsky, K., Bryant, L. D., and Wallmann, K.: Organic carbon production, mineralisation and preservation on the Peruvian margin, Biogeosciences, 12, 1537–1559, https://doi.org/10.5194/bg-12-1537-2015, 2015.
Davies, P. J. and Bubela, B.: The transformation of nesquehonite into hydromagnesite, Chem. Geol., 12, 289–300, https://doi.org/10.1016/0009-2541(73)90006-5, 1973.
de Lannoy, C.-F., Eisaman, M. D., Jose, A., Karnitz, S. D., DeVaul, R. W., Hannun, K., and Rivest, J. L. B.: Indirect ocean capture of atmospheric CO2: Part I. Prototype of a negative emissions technology, Int. J. Greenh. Gas Con., 70, 243–253, https://doi.org/10.1016/j.ijggc.2017.10.007, 2018.
Doelman, C. J., Leurs, R., Oosterom, W. C., and Bast, A.: Mineral dust exposure and free radical-mediated lung damage, Exp. Lung Res., 16, 41–55, https://doi.org/10.3109/01902149009064698, 1990.
Du, L., Song, Q., and Jia, X.: Detecting concept drift: an information entropy-based method using an adaptive sliding window, Intell. Data Anal., 18, 337–364, 2014.
Edwards, H., Moody, C., Newton, E., Jorge-Villar, S., and Russell, M.: Raman Spectroscopic analysis of cyanobacterial colonisation of hydromagnesite, a putative Martian extremophile, Icarus, 175, 372–381, https://doi.org/10.1016/j.icarus.2004.12.006, 2005.
Eisaman, M. D.: Negative Emissions Technologies: The Tradeoffs of Air-Capture Economics, Joule, 4, 516–520, https://doi.org/10.1016/j.joule.2020.02.007, 2020.
Eisaman, M. D., Parajuly, K., Tuganov, A., Eldershaw, C., Chang, N., and Littau, K. A.: CO2 extraction from seawater using bipolar membrane electrodialysis, Energy Environ. Sci., 5, 7346–7352, https://doi.org/10.1039/c2ee03393c, 2012.
Eisaman, M. D., Rivest, J. L. B., Karnitz, S. D., de Lannoy, C.-F., Jose, A., DeVaul, R. W., and Hannun, K.: Indirect ocean capture of atmospheric CO2: Part II. Understanding the cost of negative emissions, Int. J. Greenh. Gas Con., 70, 254–261, https://doi.org/10.1016/j.ijggc.2018.02.020, 2018.
European Commission: Joint Research Centre. Institute for Prospective Technological Studies: Best available techniques (BAT) reference document for the production of cement, lime and magnesium oxide: Industrial Emissions Directive 2010/75/EU (integrated pollution prevention and control), Publications Office, LU, https://op.europa.eu/en/publication-detail/-/publication/12dbe9f3-28c6-44c9-8962-50a1359443d6 (last access: 6 November 2023), 2013.
European Commission: Commission Decision (EU) 2017/848 – of 17 May 2017 – laying down criteria and methodological standards on good environmental status of marine waters and specifications and standardised methods for monitoring and assessment, and repealing Decision 2010/ 477/ EU, https://eur-lex.europa.eu/resource.html?uri=cellar:ef454a92-98a9-11e7-b92d-01aa75ed71a1.0013.02/DOC_1&format=PDF (last access: 6 November 2023), 2017.
Fakhraee, M., Li, Z., Planavsky, N. J., and Reinhard, C. T.: A biogeochemical model of mineral-based ocean alkalinity enhancement: impacts on the biological pump and ocean carbon uptake, Environ. Res. Lett., 18, 044047, https://doi.org/10.1088/1748-9326/acc9d4, 2023.
Fischer, H. C.: Calcination of Calcite: I, Effect of Heating Rate and Temperature on Bulk Density of Calcium Oxide, J. Am. Ceram. Soc., 38, 245–251, https://doi.org/10.1111/j.1151-2916.1955.tb14939.x, 1955.
Flipkens, G., Blust, R., and Town, R. M.: Deriving Nickel (Ni(II)) and Chromium (Cr(III)) Based Environmentally Safe Olivine Guidelines for Coastal Enhanced Silicate Weathering, Environ. Sci. Technol., 55, 12362–12371, https://doi.org/10.1021/acs.est.1c02974, 2021.
Flipkens, G., Fuhr, M., Fiers, G., Meysman, F. J. R., Town, R. M., and Blust, R.: Enhanced olivine dissolution in seawater through continuous grain collisions, Geochim. Cosmochim. Ac., 359, 84–99, https://doi.org/10.1016/j.gca.2023.09.002, 2023.
Foteinis, S., Andresen, J., Campo, F., Caserini, S., and Renforth, P.: Life cycle assessment of ocean liming for carbon dioxide removal from the atmosphere, J. Clean Prod., 370, 133309, https://doi.org/10.1016/j.jclepro.2022.133309, 2022.
Foteinis, S., Campbell, J. S., and Renforth, P.: Life Cycle Assessment of Coastal Enhanced Weathering for Carbon Dioxide Removal from Air, Environ. Sci. Technol., 57, 6169–6178, https://doi.org/10.1021/acs.est.2c08633, 2023.
Fuhr, M., Geilert, S., Schmidt, M., and Wallmann, K.: Kinetics of olivine weathering in seawater: an experimental study, in: Goldschmidt2021 abstracts, Goldschmidt2021, Virtual, https://doi.org/10.7185/gold2021.7375, 2021.
Garai, P., Banerjee, P., Mondal, P., and Saha, N. C.: Effect of Heavy Metals on Fishes: Toxicity and Bioaccumulation, J. Clin. Toxicol., S18, 001, https://www.longdom.org/open-access/effect-of-heavy-metals-on-fishes-toxicity-and-bioaccumulation-82260.html (last access: 15 November 2023), 2021.
Gartner, E.: Discussion of the paper “A new view on the kinetics of tricalcium silicate hydration,” by L. Nicoleau and A. Nonat, Cem. Concr. Res. 86 (2016) 1–11, Cement Concrete Res., 104, 114–117, https://doi.org/10.1016/j.cemconres.2017.10.015, 2018.
Gattuso, J.-P., Frankignoulle, M., and Wollast, R.: Carbon and Carbonate Metabolism in Coastal Aquatic Ecosystems, Annu. Rev. Ecol. Syst., 29, 405–434, https://doi.org/10.1146/annurev.ecolsys.29.1.405, 1998.
Geilert, S., Frick, D. A., Garbe-Schönberg, D., Scholz, F., Sommer, S., Grasse, P., Vogt, C., and Dale, A. W.: Coastal El Niño triggers rapid marine silicate alteration on the seafloor, Nat. Commun., 14, 1676, https://doi.org/10.1038/s41467-023-37186-5, 2023.
Glass, J. B. and Dupont, C. L.: Oceanic Nickel Biogeochemistry and the Evolution of Nickel Use, in: The Biological Chemistry of Nickel, edited by: Zamble, D., Rowińska-Żyrek, M., and Kozlowski, H., The Royal Society of Chemistry, 12–26, https://doi.org/10.1039/9781788010580-00012, 2017.
Gore, S., Renforth, P., and Perkins, R.: The potential environmental response to increasing oceanalkalinity for negative emissions, Mitig. Adapt. Strateg. Glob. Change, 24, 1191–1211, https://doi.org/10.1007/s11027-018-9830-z, 2019.
Greinert, J. and Derkachev, A.: Glendonites and methane-derived Mg-calcites in the Sea of Okhotsk, Eastern Siberia: implications of a venting-related ikaite/glendonite formation, Mar. Geol., 204, 129–144, https://doi.org/10.1016/S0025-3227(03)00354-2, 2004.
Guo, J. A., Strzepek, R., Willis, A., Ferderer, A., and Bach, L. T.: Investigating the effect of nickel concentration on phytoplankton growth to assess potential side-effects of ocean alkalinity enhancement, Biogeosciences, 19, 3683–3697, https://doi.org/10.5194/bg-19-3683-2022, 2022.
Guy, C. and Schott, J.: Multisite surface reaction versus transport control during the hydrolysis of a complex oxide, Chem. Geol., 78, 181–204, https://doi.org/10.1016/0009-2541(89)90057-0, 1989.
Haas, S., Weber, N., Berry, A., and Erich, E.: Limestone powder carbon dioxide scrubber as the technology for Carbon Capture and Usage, Cement Int., 3, 34−-45, 2014.
Hamilton, J. L., Wilson, S. A., Morgan, B., Harrison, A. L., Turvey, C. C., Paterson, D. J., Dipple, G. M., and Southam, G.: Accelerating Mineral Carbonation in Ultramafic Mine Tailings via Direct CO2 Reaction and Heap Leaching with Potential for Base Metal Enrichment and Recovery, Econ. Geol., 115, 303–323, https://doi.org/10.5382/econgeo.4710, 2020.
Hangx, S. J. T. and Spiers, C. J.: Coastal spreading of olivine to control atmospheric CO2 concentrations: A critical analysis of viability, Int. J. Greenh. Gas Con., 3, 757–767, https://doi.org/10.1016/j.ijggc.2009.07.001, 2009.
Hartmann, J. and Moosdorf, N.: The new global lithological map database GLiM: A representation of rock properties at the Earth surface: Technical Brief, Geochem. Geophy. Geosy., 13, Q12004, https://doi.org/10.1029/2012GC004370, 2012.
Hartmann, J., Jansen, N., Dürr, H. H., Kempe, S., and Köhler, P.: Global CO2-consumption by chemical weathering: What is the contribution of highly active weathering regions?, Global Planet. Change, 69, 185–194, https://doi.org/10.1016/j.gloplacha.2009.07.007, 2009.
Hartmann, J., Suitner, N., Lim, C., Schneider, J., Marín-Samper, L., Arístegui, J., Renforth, P., Taucher, J., and Riebesell, U.: Stability of alkalinity in ocean alkalinity enhancement (OAE) approaches – consequences for durability of CO2 storage, Biogeosciences, 20, 781–802, https://doi.org/10.5194/bg-20-781-2023, 2023.
Harvey, L. D. D.: Mitigating the atmospheric CO2 increase and ocean acidification by adding limestone powder to upwelling regions, J. Geophys. Res., 113, C04028, https://doi.org/10.1029/2007JC004373, 2008.
Hauck, J., Köhler, P., Wolf-Gladrow, D., and Völker, C.: Iron fertilisation and century-scale effects of open ocean dissolution of olivine in a simulated CO2 removal experiment, Environ. Res. Lett., 11, 024007, https://doi.org/10.1088/1748-9326/11/2/024007, 2016.
He, J. and Tyka, M. D.: Limits and CO2 equilibration of near-coast alkalinity enhancement, Biogeosciences, 20, 27–43, https://doi.org/10.5194/bg-20-27-2023, 2023.
Heder, M.: From NASA to EU: the evolution of the TRL scale in Public Sector Innovation, The Innovation Journal, 22, 3, https://web.archive.org/web/20171011071816/https://www.innovation.cc/discussion-papers/22_2_3_heder_nasa-to-eu-trl-scale.pdf (last access: 17 November 2023), 2017.
Hinga, K.: Effects of pH on coastal marine phytoplankton, Mar. Ecol. Prog. Ser., 238, 281–300, https://doi.org/10.3354/meps238281, 2002.
Ho, D. T., Bopp, L., Palter, J. B., Long, M. C., Boyd, P. W., Neukermans, G., and Bach, L. T.: Monitoring, reporting, and verification for ocean alkalinity enhancement, in: Guide to Best Practices in Ocean Alkalinity Enhancement Research, edited by: Oschlies, A., Stevenson, A., Bach, L. T., Fennel, K., Rickaby, R. E. M., Satterfield, T., Webb, R., and Gattuso, J.-P., Copernicus Publications, State Planet, 2-oae2023, 12, https://doi.org/10.5194/sp-2-oae2023-12-2023, 2023.
Hopkinson, L., Rutt, K., and Cressey, G.: The transformation of nesquehonite to hydromagnesite in the system CaO-MgO-H2O-CO2: An experimental spectroscopic study, J. Geol., 116, 387–400, 2008.
House, K. Z., House, C. H., Schrag, D. P., and Aziz, M. J.: Electrochemical Acceleration of Chemical Weathering as an Energetically Feasible Approach to Mitigating Anthropogenic Climate Change, Environ. Sci. Technol., 41, 8464–8470, https://doi.org/10.1021/es0701816, 2007.
Hu, Y.-B., Wolf-Gladrow, D. A., Dieckmann, G. S., Völker, C., and Nehrke, G.: A laboratory study of ikaite (CaCO3 • 6H2O) precipitation as a function of pH, salinity, temperature and phosphate concentration, Mar. Chem., 162, 10–18, https://doi.org/10.1016/j.marchem.2014.02.003, 2014.
Huntington, S.: A guide to using calcium reactors, Reefkeeping Online Magazine, http://www.reefkeeping.com/issues/2002-05/sh/feature/ (last access: 17 November 2023), 2002.
Ilyina, T., Wolf-Gladrow, D., Munhoven, G., and Heinze, C.: Assessing the potential of calcium-based artificial ocean alkalinization to mitigate rising atmospheric CO2 and ocean acidification, Geophys. Res. Lett., 40, 5909–5914, https://doi.org/10.1002/2013GL057981, 2013.
ISO: Environmental management: life cycle assessment: principles and framework, Vol. ISO 14040, https://www.iso.org/standard/37456.html (last access: 15 November 23), 2006.
Jahnke, R. A. and Jahnke, D. B.: Rates of C, N, P and Si recycling and denitrification at the US Mid-Atlantic continental slope depocenter, Deep-Sea Res. Pt. I, 47, 1405–1428, https://doi.org/10.1016/S0967-0637(99)00118-1, 2000.
Jeandel, C. and Oelkers, E. H.: The influence of terrigenous particulate material dissolution on ocean chemistry and global element cycles, Chem. Geol., 395, 50–66, https://doi.org/10.1016/j.chemgeo.2014.12.001, 2015.
Jeandel, C., Peucker-Ehrenbrink, B., Jones, M. T., Pearce, C. R., Oelkers, E. H., Godderis, Y., Lacan, F., Aumont, O., and Arsouze, T.: Ocean margins: The missing term in oceanic element budgets?, Eos Trans. AGU, 92, 217–218, https://doi.org/10.1029/2011EO260001, 2011.
Kazakov, A. V., Tikhomirova, M. M., and Plotnikova, V. I.: The System of Carbonate Equilibria, Int. Geol. Rev., 1, 1–39, https://doi.org/10.1080/00206815909473450, 1959.
Kelland, M., Rau, G., Battochio, B., Vallis, J., Gladkovas, M., Thomas, S., Bradley, K., Brereton, C., Garg, S., and Mezei, A.: Integrating Carbon Capture in Mining Through Metallurgy. Part 1: Leaching and Reclamation of Asbestos Tailings: Thetford Mines Carbon Capture and Remediation Project, in: Proceedings of the 61st Conference of Metallurgists, COM 2022, COM 2022, Springer, Cham, 515–527, https://doi.org/10.1007/978-3-031-17425-4_66, 2022.
Kheshgi, H. S.: Sequestering atmospheric carbon dioxide by increasing ocean alkalinity, Energy, 20, 915–922, https://doi.org/10.1016/0360-5442(95)00035-F, 1995.
Kim, S., Nitzsche, M. P., Rufer, S. B., Lake, J. R., Varanasi, K. K., and Hatton, T. A.: Asymmetric chloride-mediated electrochemical process for CO2 removal from oceanwater, Energy Environ. Sci., 16, 2030–2044, https://doi.org/10.1039/D2EE03804H, 2023.
Kim, Y., Caumon, M.-C., Barres, O., Sall, A., and Cauzid, J.: Identification and composition of carbonate minerals of the calcite structure by Raman and infrared spectroscopies using portable devices, Spectrochim. Acta A, 261, 119980, https://doi.org/10.1016/j.saa.2021.119980, 2021.
Kirchner, J. S., Lettmann, K. A., Schnetger, B., Wolff, J.-O., and Brumsack, H.-J.: Carbon capture via accelerated weathering of limestone: Modeling local impacts on the carbonate chemistry of the southern North Sea, Int. J. Greenh. Gas Con., 92, 102855, https://doi.org/10.1016/j.ijggc.2019.102855, 2020a.
Kirchner, J. S., Berry, A., Ohnemüller, F., Schnetger, B., Erich, E., Brumsack, H.-J., and Lettmann, K. A.: Reducing CO 2 Emissions of a Coal-Fired Power Plant via Accelerated Weathering of Limestone: Carbon Capture Efficiency and Environmental Safety, Environ. Sci. Technol., 54, 4528–4535, https://doi.org/10.1021/acs.est.9b07009, 2020b.
Kirschvink, J. L.: Late Proterozoic Low-Latitude Global Glaciation: The Snowball Earth, in: the Proterozoic Biosphere: A Multidisciplinary Study, edited by: Schopf, J. and Klein, C., 51–52, Cambridge University Press, Cambridge, UK, ISBN 9780521366151, 1992.
Köhler, P., Abrams, J. F., Völker, C., Hauck, J., and Wolf-Gladrow, D. A.: Geoengineering impact of open ocean dissolution of olivine on atmospheric CO2, surface ocean pH and marine biology, Environ. Res. Lett., 8, 014009, https://doi.org/10.1088/1748-9326/8/1/014009, 2013.
Königsberger, E., Königsberger, L.-C., and Gamsjäger, H.: Low-temperature thermodynamic model for the system Na2CO3–MgCO3–CaCO3–H2O, Geochim. Cosmochim. Ac., 63, 3105–3119, https://doi.org/10.1016/S0016-7037(99)00238-0, 1999.
Kumar, A., Du, F., and Lienhard, J. H.: Caustic Soda Production, Energy Efficiency, and Electrolyzers, ACS Energy Lett., 6, 3563–3566, https://doi.org/10.1021/acsenergylett.1c01827, 2021.
La Plante, E. C., Simonetti, D. A., Wang, J., Al-Turki, A., Chen, X., Jassby, D., and Sant, G. N.: Saline Water-Based Mineralization Pathway for Gigatonne-Scale CO2 Management, ACS Sustainable Chem. Eng., 9, 1073–1089, https://doi.org/10.1021/acssuschemeng.0c08561, 2021.
La Plante, E. C., Chen, X., Bustillos, S., Bouissonnie, A., Traynor, T., Jassby, D., Corsini, L., Simonetti, D. A., and Sant, G. N.: Electrolytic Seawater Mineralization and the Mass Balances That Demonstrate Carbon Dioxide Removal, ACS EST Eng., 3, 955–968, https://doi.org/10.1021/acsestengg.3c00004, 2023.
Lammers, L. N., Duan, Y., Anaya, L., Koishi, A., Lopez, R., Delima, R., Jassby, D., and Sedlak, D. L.: Electrolytic Sulfuric Acid Production with Carbon Mineralization for Permanent Carbon Dioxide Removal, ACS Sustainable Chem. Eng., 11, 4800–4812, https://doi.org/10.1021/acssuschemeng.2c07441, 2023.
Langer, W. H., San Juan, C. A., Rau, G. H. and Caldeira, K.: Accelerated weathering of limestone for CO2 mitigation: Opportunities for the stone and cement industries, Mining. Eng., 61, 27–32, https://www.researchgate.net/publication/283868780_Accelerated_weathering_of limestone_for_CO2_mitigation_Opportunities_for_the_stone_and_cement_industries#fullTextFileContent (last access: 6 November 2023), 2009.
Laruelle, G. G., Lauerwald, R., Pfeil, B., and Regnier, P.: Regionalized global budget of the CO2 exchange at the air-water interface in continental shelf seas: Continental shelf seas CO2 fluxes, Global Biogeochem. Cy., 28, 1199–1214, https://doi.org/10.1002/2014GB004832, 2014.
Laruelle, G. G., Cai, W.-J., Hu, X., Gruber, N., Mackenzie, F. T., and Regnier, P.: Continental shelves as a variable but increasing global sink for atmospheric carbon dioxide, Nat. Commun., 9, 454, https://doi.org/10.1038/s41467-017-02738-z, 2018.
Lennie, A. R., Tang, C. C., and Thompson, S. P.: The structure and thermal expansion behaviour of ikaite, CaCO3 • 6H2O, from T=114 to T=293 K, Mineral. Mag., 68, 135–146, https://doi.org/10.1180/0026461046810176, 2004.
Lewis, E. and Wallace, D. W. R.: Program Developed for CO2 System Calculations, CDIAC, ESS-DIVE repository [data set], https://doi.org/10.15485/1464255, 1998.
Linke, P., Suess, E., Torres, M., Martens, V., Rugh, W. D., Ziebis, W., and Kulm, L. D.: In situ measurement of fluid flow from cold seeps at active continental margins, Deep-Sea Res. Pt. I, 41, 721–739, https://doi.org/10.1016/0967-0637(94)90051-5, 1994.
Longhurst, A., Sathyendranath, S., Platt, T., and Caverhill, C.: An estimate of global primary production in the ocean from satellite radiometer data, J. Plankton Res., 17, 1245–1271, https://doi.org/10.1093/plankt/17.6.1245, 1995.
Madeddu, S., Priestnall, M., Godoy, E., Kumar, R. V., Raymahasay, S., Evans, M., Wang, R., Manenye, S., and Kinoshita, H.: Extraction of Mg(OH)2 from Mg silicate minerals with NaOH assisted with H2O: implications for CO2 capture from exhaust flue gas, Faraday Discuss., 183, 369–387, https://doi.org/10.1039/C5FD00047E, 2015.
Marion, G. M.: Carbonate mineral solubility at low temperatures in the Na–K–Mg–Ca–H–Cl–SO4–OH–HCO3–CO3–CO2–H2O system, Geochim. Cosmochim. Ac., 65, 1883–1896, https://doi.org/10.1016/S0016-7037(00)00588-3, 2001.
Marland, G.: Phase equilibria in the system calcium carbonate-water, Geochim. Cosmochim. Ac., 39, 1193–1197, https://doi.org/10.1016/0016-7037(75)90061-7, 1975.
McLaren, D.: A comparative global assessment of potential negative emissions technologies, Process. Saf. Environ., 90, 489–500, https://doi.org/10.1016/j.psep.2012.10.005, 2012.
McNeil, B. I., Matear, R. J., Key, R. M., Bullister, J. L., and Sarmiento, J. L.: Anthropogenic CO2 Uptake by the Ocean Based on the Global Chlorofluorocarbon Data Set, Science, 299, 235–239, https://doi.org/10.1126/science.1077429, 2003.
McQueen, N., Kelemen, P., Dipple, G., Renforth, P., and Wilcox, J.: Ambient weathering of magnesium oxide for CO2 removal from air, Nat. Commun., 11, 3299, https://doi.org/10.1038/s41467-020-16510-3, 2020.
Meysman, F. J. R. and Montserrat, F.: Negative CO2 emissions via enhanced silicate weathering in coastal environments, Biol. Lett., 13, 20160905, https://doi.org/10.1098/rsbl.2016.0905, 2017.
Meysman, F. J. R., Cornelissen, R., Trashin, S., Bonné, R., Martinez, S. H., Van Der Veen, J., Blom, C. J., Karman, C., Hou, J.-L., Eachambadi, R. T., Geelhoed, J. S., Wael, K. D., Beaumont, H. J. E., Cleuren, B., Valcke, R., Van Der Zant, H. S. J., Boschker, H. T. S., and Manca, J. V.: A highly conductive fibre network enables centimetre-scale electron transport in multicellular cable bacteria, Nat. Commun., 10, 4120, https://doi.org/10.1038/s41467-019-12115-7, 2019.
Middelburg, J. J., Soetaert, K., and Hagens, M.: Ocean alkalinity, buffering and biogeochemical processes, Rev. Geophys., 58, e2019RG000681, https://doi.org/10.1029/2019RG000681, 2020.
Mikkelsen, A., Andersen, A. B., Engelsen, S. B., Hansen, H. C. B., Larsen, O., and Skibsted, L. H.: Presence and Dehydration of Ikaite, Calcium Carbonate Hexahydrate, in Frozen Shrimp Shell, J. Agric. Food Chem., 47, 911–917, https://doi.org/10.1021/jf980932a, 1999.
Montserrat, F., Renforth, P., Hartmann, J., Leermakers, M., Knops, P., and Meysman, F. J. R.: Olivine Dissolution in Seawater: Implications for CO2 Sequestration through Enhanced Weathering in Coastal Environments, Environ. Sci. Technol., 51, 3960–3972, https://doi.org/10.1021/acs.est.6b05942, 2017.
Moras, C. A., Bach, L. T., Cyronak, T., Joannes-Boyau, R., and Schulz, K. G.: Ocean alkalinity enhancement – avoiding runaway CaCO3 precipitation during quick and hydrated lime dissolution, Biogeosciences, 19, 3537–3557, https://doi.org/10.5194/bg-19-3537-2022, 2022.
Moropoulou, A., Bakolas, A., and Aggelakopoulou, E.: The Effects of Limestone Characteristics and Calcination Temperature on the Reactivity of Quicklime, Cement Concrete Res., 31, 633–639, https://doi.org/10.1016/S0008-8846(00)00490-7, 2001.
Nduagu, E., Björklöf, T., Fagerlund, J., Wärnå, J., Geerlings, H., and Zevenhoven, R.: Production of magnesium hydroxide from magnesium silicate for the purpose of CO2 mineralisation – Part 1: Application to Finnish serpentinite, Miner. Eng., 30, 75–86, https://doi.org/10.1016/j.mineng.2011.12.004, 2012.
Neumann, A., Van Beusekom, J. E. E., Eisele, A., Emeis, K., Friedrich, J., Kröncke, I., Logemann, E. L., Meyer, J., Naderipour, C., Schückel, U., Wrede, A., and Zettler, M. L.: Macrofauna as a major driver of bentho-pelagic exchange in the southern North Sea, Limnol. Oceanogr., 66, 2203–2217, https://doi.org/10.1002/lno.11748, 2021.
Nishiyama, R., Munemoto, T., and Fukushi, K.: Formation condition of monohydrocalcite from CaCl2–MgCl2–Na2CO3 solutions, Geochim. Cosmochim. Ac., 100, 217–231, https://doi.org/10.1016/j.gca.2012.09.002, 2013.
O'Brien, T., Bommaraju, T. V., and Hine, F.: Handbook of chlor-alkali technology, Springer, New York, 5 pp., https://doi.org/10.1007/b113786, 2005.
Oelkers, E. H., Declercq, J., Saldi, G. D., Gislason, S. R., and Schott, J.: Olivine dissolution rates: A critical review, Chem. Geol., 500, 1–19, https://doi.org/10.1016/j.chemgeo.2018.10.008, 2018.
Orr, J. C., Fabry, V. J., Aumont, O., Bopp, L., Doney, S. C., Feely, R. A., Gnanadesikan, A., Gruber, N., Ishida, A., Joos, F., Key, R. M., Lindsay, K., Maier-Reimer, E., Matear, R., Monfray, P., Mouchet, A., Najjar, R. G., Plattner, G.-K., Rodgers, K. B., Sabine, C. L., Sarmiento, J. L., Schlitzer, R., Slater, R. D., Totterdell, I. J., Weirig, M.-F., Yamanaka, Y., and Yool, A.: Anthropogenic ocean acidification over the twenty-first century and its impact on calcifying organisms, Nature, 437, 681–686, https://doi.org/10.1038/nature04095, 2005.
Oskierski, H. C., Turvey, C. C., Wilson, S., Dlugogorski, B. Z., Altarawneh, M., and Mavromatis, V.: Mineralisation of atmospheric CO2 in hydromagnesite in ultramafic mine tailings – Insights from Mg isotopes, Geochim. Cosmochim. Ac., 309, 191–208, https://doi.org/10.1016/j.gca.2021.06.020, 2021.
Pan, X.-J., Dou, Z.-H., Zhang, T.-A., Meng, D.-L., and Fan, Y.-Y.: Separation of metal ions and resource utilization of magnesium from saline lake brine by membrane electrolysis, Sep. Purif. Technol., 251, 117316, https://doi.org/10.1016/j.seppur.2020.117316, 2020.
Papadimitriou, S., Kennedy, H., Kennedy, P., and Thomas, D. N.: Kinetics of ikaite precipitation and dissolution in seawater-derived brines at sub-zero temperatures to 265 K, Geochim. Cosmochim. Ac., 140, 199–211, https://doi.org/10.1016/j.gca.2014.05.031, 2014.
Pedersen, F. M. and Hansen, P.: Effects of high pH on natural planktonic community, Mar. Ecol.-Prog. Ser., 260, 19–31, https://doi.org/10.3354/meps260019, 2003.
Penman, D. E. and Rooney, A. D.: Coupled carbon and silica cycle perturbations during the Marinoan snowball Earth deglaciation, Geology, 47, 317–320, https://doi.org/10.1130/G45812.1, 2019.
Pesce, C., Godina, M. C., Henry, A., and Pesce, G. L.: Effects of Steam-Slaking on the Characteristics of Lime from Three Different UK Manufacturers, in: Proceedings of the 75th RILEM Annual Week 2021, Merida, Mexico, 29 August–3 September 2021, Springer Cham, 771–780, https://doi.org/10.1007/978-3-031-21735-7_82, 2023.
Petit, J. R., Jouzel, J., Raynaud, D., Barkov, N. I., Delaygue, G., Delmotte, M., Kotlyakov, V. M., Legrand, M., Lipenkov, V. Y., Lorius, C., and Saltzman, E.: Climate and atmospheric history of the past 420,000 years from the Vostok ice core, Antarctica, Nature 399, 429–436, https://doi.org/10.1038/20859, 1999.
Pollyea, R. M. and Rimstidt, J. D.: Rate equations for modeling carbon dioxide sequestration in basalt, Appl. Geochem., 81, 53–62, https://doi.org/10.1016/j.apgeochem.2017.03.020, 2017.
Rahman, S., Aller, R. C., and Cochran, J. K.: The Missing Silica Sink: Revisiting the Marine Sedimentary Si Cycle Using Cosmogenic 32Si: The Missing Sedimentary Silica Sink, Global Biogeochem. Cy., 31, 1559–1578, https://doi.org/10.1002/2017GB005746, 2017.
Rau, G. H.: Electrochemical Splitting of Calcium Carbonate to Increase Solution Alkalinity: Implications for Mitigation of Carbon Dioxide and Ocean Acidity, Environ. Sci. Technol., 42, 8935–8940, https://doi.org/10.1021/es800366q, 2008.
Rau, G. H.: CO2 Mitigation via Capture and Chemical Conversion in Seawater, Environ. Sci. Technol., 45, 1088–1092, https://doi.org/10.1021/es102671x, 2011.
Rau, G. H. and Caldeira, K.: Enhanced carbonate dissolution: A means of sequestering waste CO2 as ocean bicarbonate, Energ. Convers. Manag., 40, 1803–1813, https://doi.org/10.1016/S0196-8904(99)00071-0, 1999.
Rau, G. H., Knauss, K. G., Langer, W. H., and Caldeira, K.: Reducing energy-related CO2 emissions using accelerated weathering of limestone, Energy, 32, 1471–1477, https://doi.org/10.1016/j.energy.2006.10.011, 2007.
Rau, G. H., Carroll, S. A., Bourcier, W. L., Singleton, M. J., Smith, M. M., and Aines, R. D.: Direct electrolytic dissolution of silicate minerals for air CO2 mitigation and carbon-negative H2 production, P. Natl. Acad. Sci. USA, 110, 10095–10100, https://doi.org/10.1073/pnas.1222358110, 2013.
Rau, G., Rackley, S., Burt, W., Tahmasebi, S., Sadoon, O., Kielly, B., and Kelland, M.: Initial Outline of OAE CDR MRV: Concepts and Needs, Planetary Technologies, Inc., https://www.planetarytech.com/wp-content/uploads/2022/03/OAE-MRV-Concepts.pdf (last access: 17 November 2023), 2022.
Regnier, P., Friedlingstein, P., Ciais, P., Mackenzie, F. T., Gruber, N., Janssens, I. A., Laruelle, G. G., Lauerwald, R., Luyssaert, S., Andersson, A. J., Arndt, S., Arnosti, C., Borges, A. V., Dale, A. W., Gallego-Sala, A., Goddéris, Y., Goossens, N., Hartmann, J., Heinze, C., Ilyina, T., Joos, F., LaRowe, D. E., Leifeld, J., Meysman, F. J. R., Munhoven, G., Raymond, P. A., Spahni, R., Suntharalingam, P., and Thullner, M.: Anthropogenic perturbation of the carbon fluxes from land to ocean, Nat. Geosci., 6, 597–607, https://doi.org/10.1038/ngeo1830, 2013.
Renforth, P. and Campbell, J. S.: The role of soils in the regulation of ocean acidification, Phil. Trans. R. Soc. B, 376, 20200174, https://doi.org/10.1098/rstb.2020.0174, 2021.
Renforth, P. and Henderson, G.: Assessing ocean alkalinity for carbon sequestration: Ocean Alkalinity for C Sequestration, Rev. Geophys., 55, 636–674, https://doi.org/10.1002/2016RG000533, 2017.
Renforth, P. and Kruger, T.: Coupling Mineral Carbonation and Ocean Liming, Energy Fuels, 27, 4199–4207, https://doi.org/10.1021/ef302030w, 2013.
Renforth, P., Jenkins, B. G., and Kruger, T.: Engineering challenges of ocean liming, Energy, 60, 442–452, https://doi.org/10.1016/j.energy.2013.08.006, 2013.
Renforth, P., Baltruschat, S., Peterson, K., Mihailova, B. D., and Hartmann, J.: Using ikaite and other hydrated carbonate minerals to increase ocean alkalinity for carbon dioxide removal and environmental remediation, Joule, 6, 2674–2679, https://doi.org/10.1016/j.joule.2022.11.001, 2022.
Riebesell, U., Basso, D., Geilert, S., Dale, A. W., and Kreuzburg, M.: Mesocosm experiments in ocean alkalinity enhancement research, in: Guide to Best Practices in Ocean Alkalinity Enhancement Research, edited by: Oschlies, A., Stevenson, A., Bach, L. T., Fennel, K., Rickaby, R. E. M., Satterfield, T., Webb, R., and Gattuso, J.-P., Copernicus Publications, State Planet, 2-oae2023, 6, https://doi.org/10.5194/sp-2-oae2023-6-2023, 2023.
Rigopoulos, I., Harrison, A. L., Delimitis, A., Ioannou, I., Efstathiou, A. M., Kyratsi, T., and Oelkers, E. H.: Carbon sequestration via enhanced weathering of peridotites and basalts in seawater, Appl. Geochem., 91, 197–207, https://doi.org/10.1016/j.apgeochem.2017.11.001, 2018.
Rodriguez-Blanco, J. D., Shaw, S., and Benning, L. G.: The kinetics and mechanisms of amorphous calcium carbonate (ACC) crystallization to calcite, viavaterite, Nanoscale, 3, 265–271, https://doi.org/10.1039/C0NR00589D, 2011.
Russell, M. J., Keith, J. I., Veysel, Z., Derya, M., Filiz, S., Allan, J. H., and Anthony, E. F.: Search for signs of ancient life on Mars: Expectations from hydromagnesite microbialites, Salda Lake, Turkey, Journal of the Geological Society, 156, 869–888, https://doi.org/10.1144/gsjgs.156.5.0869, 1999.
Sano, Y., Hao, Y., and Kuwahara, F.: Development of an electrolysis based system to continuously recover magnesium from seawater, Heliyon, 4, e00923, https://doi.org/10.1016/j.heliyon.2018.e00923, 2018.
Sarmiento, J. L. and Gruber, N.: Ocean Biogeochemical Dynamics, Princeton University Press, ISBN 9780691017075, 2006.
Schuiling, R. D. and de Boer, P. L.: Coastal spreading of olivine to control atmospheric CO2 concentrations: A critical analysis of viability. Comment: Nature and laboratory models are different, Int. J. Greenh. Gas Con., 4, 855–856, 2010.
Schuiling, R. D. and de Boer, P. L.: Rolling stones; fast weathering of olivine in shallow seas for cost-effective CO2 capture and mitigation of global warming and ocean acidification, Earth Syst. Dynam. Discuss., 2, 551–568, https://doi.org/10.5194/esdd-2-551-2011, 2011.
Schulz, H. D. and Zabel, M.: Marine Geochemistry, 2nd edn., 574 pp., Springer Berlin, https://doi.org/10.1007/3-540-32144-6, 2006.
Schulz, K. G., Bach, L. T., and Dickson, A. G.: Seawater carbonate chemistry considerations for ocean alkalinity enhancement research: theory, measurements, and calculations, in: Guide to Best Practices in Ocean Alkalinity Enhancement Research, edited by: Oschlies, A., Stevenson, A., Bach, L. T., Fennel, K., Rickaby, R. E. M., Satterfield, T., Webb, R., and Gattuso, J.-P., Copernicus Publications, State Planet, 2-oae2023, 2, https://doi.org/10.5194/sp-2-oae2023-2-2023, 2023.
Seeländer, K.: Alkalinity enhancement in rivers, Bachelor Thesis, University of Southern Denmark, Odense, DK, 2023.
Shahar, A., Bassett, W. A., Mao, H.-K., Chou, I.-M., and Mao, W.: The stability and Raman spectra of ikaite, CaCO3 • 6H2O, at high pressure and temperature, 90, 1835–1839, https://doi.org/10.2138/am.2005.1783, 2005.
Simkin, T. and Smith, J. V.: Minor-Element Distribution in Olivine, J. Geol., 78, 304–325, https://doi.org/10.1086/627519, 1970.
Smith, S. M., Geden, O., Nemet, G. F., Gidden, M. J., Lamb, W. F., Powis, C., Bellamy, R., Callaghan, M. W., Cowie, A., Cox, E., Fuss, S., Gasser, T., Grassi, G., Greene, J., Lück, S., Mohan, A., Müller-Hansen, F., Peters, G. P., Pratama, Y., Repke, T., Riahi, K., Schenuit, F., Steinhauser, J., Strefler, J., Valenzuela, J. M., and Minx, J. C.: The State of Carbon Dioxide Removal, 1st edn., The State of Carbon Dioxide Removal, https://doi.org/10.17605/OSF.IO/W3B4Z, 2023
Sommer, S., Gier, J., Treude, T., Lomnitz, U., Dengler, M., Cardich, J., and Dale, A. W.: Depletion of oxygen, nitrate and nitrite in the Peruvian oxygen minimum zone cause an imbalance of benthic nitrogen fluxes, Deep-Sea Res. Pt. I, 112, 113–122, https://doi.org/10.1016/j.dsr.2016.03.001, 2016.
Sonnichsen, C., Atamanchuk, D., Hendricks, A., Morgan, S., Smith, J., Grundke, I., Luy, E., and Sieben, V. J.: An Automated Microfluidic Analyzer for In Situ Monitoring of Total Alkalinity, ACS Sens., 8, 344–352, https://doi.org/10.1021/acssensors.2c02343, 2023.
Stolaroff, J. K., Keith, D. W., and Lowry, G. V.: Carbon Dioxide Capture from Atmospheric Air Using Sodium Hydroxide Spray, Environ. Sci. Technol., 42, 2728–2735, https://doi.org/10.1021/es702607w, 2008.
Strathmann, H., Giorno, L., and Drioli, E.: An Introduction to Membrane Science and Technology, Inst. on Membrane Technology, 394 pp., ISBN 9788880800637, 8880800639, 2006.
Strefler, J., Amann, T., Bauer, N., Kriegler, E., and Hartmann, J.: Potential and costs of carbon dioxide removal by enhanced weathering of rocks, Environ. Res. Lett., 13, 034010, https://doi.org/10.1088/1748-9326/aaa9c4, 2018.
Sun, X. and Turchyn, A. V.: Significant contribution of authigenic carbonate to marine carbon burial, Nat. Geosci., 7, 201–204, https://doi.org/10.1038/ngeo2070, 2014.
Talabi, O. O., Dorfi, A. E., O'Neil, G. D., and Esposito, D. V.: Membraneless electrolyzers for the simultaneous production of acid and base, Chem. Commun., 53, 8006–8009, https://doi.org/10.1039/C7CC02361H, 2017.
Tatzber, M., Stemmer, M., Spiegel, H., Katzlberger, C., Haberhauer, G., and Gerzabek, M. H.: An alternative method to measure carbonate in soils by FT-IR spectroscopy. Environ. Chem. Lett., 5, 9–12, https://doi.org/10.1007/s10311-006-0079-5, 2007.
Terlouw, T., Bauer, C., Rosa, L., and Mazzotti, M.: Life cycle assessment of carbon dioxide removal technologies: a critical review, Energy Environ. Sci., 14, 1701–1721, https://doi.org/10.1039/D0EE03757E, 2021.
Thomsen, J. K., Geest, T., and Cox, R. P.: Mass Spectrometric Studies of the Effect of pH on the Accumulation of Intermediates in Denitrification by Paracoccus denitrificans, Appl. Environ. Microbiol., 60, 536–541, https://doi.org/10.1128/aem.60.2.536-541.1994, 1994.
Torres, M. E., Hong, W.-L., Solomon, E. A., Milliken, K., Kim, J.-H., Sample, J. C., Teichert, B. M. A., and Wallmann, K.: Silicate weathering in anoxic marine sediment as a requirement for authigenic carbonate burial, Earth-Sci. Rev., 200, 102960, https://doi.org/10.1016/j.earscirev.2019.102960, 2020.
Tréguer, P. J., Sutton, J. N., Brzezinski, M., Charette, M. A., Devries, T., Dutkiewicz, S., Ehlert, C., Hawkings, J., Leynaert, A., Liu, S. M., Llopis Monferrer, N., López-Acosta, M., Maldonado, M., Rahman, S., Ran, L., and Rouxel, O.: Reviews and syntheses: The biogeochemical cycle of silicon in the modern ocean, Biogeosciences, 18, 1269–1289, https://doi.org/10.5194/bg-18-1269-2021, 2021.
Tyka, M. D., Van Arsdale, C., and Platt, J. C.: CO2 capture by pumping surface acidity to the deep ocean, Energy Environ. Sci., 15, 786–798, https://doi.org/10.1039/D1EE01532J, 2022.
Volkenborn, N., Robertson, D. M., and Reise, K.: Sediment destabilizing and stabilizing bio-engineers on tidal flats: cascading effects of experimental exclusion, Helgoland Mar. Res., 63, 27–35, https://doi.org/10.1007/s10152-008-0140-9, 2009.
Voosen, P.: Ocean geoengineering scheme aces its first field test, Science, 378, 1266–1267, https://doi.org/10.1126/science.adg3427, 2022.
Wallmann, K., Aloisi, G., Haeckel, M., Tishchenko, P., Pavlova, G., Greinert, J., Kutterolf, S., and Eisenhauer, A.: Silicate weathering in anoxic marine sediments, Geochim. Cosmochim. Ac., 72, 2895–2918, https://doi.org/10.1016/j.gca.2008.03.026, 2008.
Wallmann, K., Diesing, M., Scholz, F., Rehder, G., Dale, A. W., Fuhr, M., and Suess, E.: Erosion of carbonate-bearing sedimentary rocks may close the alkalinity budget of the Baltic Sea and support atmospheric CO2 uptake in coastal seas, Front. Mar. Sci., 9, 968069, https://doi.org/10.3389/fmars.2022.968069, 2022.
Wang, H., Pilcher, D. J., Kearney, K. A., Cross, J. N., Shugart, O. M., Eisaman, M. D., and Carter, B. R.: Simulated Impact of Ocean Alkalinity Enhancement on Atmospheric CO2 Removal in the Bering Sea, Earth's Future, 11, e2022EF002816, https://doi.org/10.1029/2022EF002816, 2023.
Willauer, H. D., DiMascio, F., Hardy, D. R., and Williams, F. W.: Feasibility of CO2 Extraction from Seawater and Simultaneous Hydrogen Gas Generation Using a Novel and Robust Electrolytic Cation Exchange Module Based on Continuous Electrodeionization Technology, Ind. Eng. Chem. Res., 53, 12192–12200, https://doi.org/10.1021/ie502128x, 2014.
Xing, L., Pullin, H., Bullock, L., Renforth, P., Darton, R. C., and Yang, A.: Potential of enhanced weathering of calcite in packed bubble columns with seawater for carbon dioxide removal, Chem. Eng. J., 431, 134096, https://doi.org/10.1016/j.cej.2021.134096, 2022.
Yakymechko, Y., Lutsyuk, I., Jaskulski, R., Dulnik, J., and Kropyvnytska, T.: The Effect of Vibro-Activation Time on the Properties of Highly Active Calcium Hydroxide, Buildings, 10, 111, https://doi.org/10.3390/buildings10060111, 2020.
Zarandi, A. E., Larachi, F., Beaudoin, G., Plante, B., and Sciortino, M.: Ambient mineral carbonation of different lithologies of mafic to ultramafic mining wastes/tailings – A comparative study, Int. J. Greenh. Gas Con., 63, 392–400, https://doi.org/10.1016/j.ijggc.2017.06.016, 2017.
- Abstract
- Introduction
- Electrochemical production of alkalinity for OAE
- Accelerated weathering of limestone as an OAE strategy
- Ocean liming
- Hydrated-carbonate-mineral formation
- Mineral addition to pelagic coastal environments
- Addition to the coastal seafloor
- Conclusions
- Appendix A: Definitions and common abbreviations
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Abstract
- Introduction
- Electrochemical production of alkalinity for OAE
- Accelerated weathering of limestone as an OAE strategy
- Ocean liming
- Hydrated-carbonate-mineral formation
- Mineral addition to pelagic coastal environments
- Addition to the coastal seafloor
- Conclusions
- Appendix A: Definitions and common abbreviations
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References